PEAKS 
蛋白质 定量
概览
为了深入了解复杂生物系统中蛋白质的功能,除了定性检测,更需要观察蛋白质的丰度变化。例如,生物标志物的发现和验证研究通常需要对蛋白质进行定量分析,以显示在不同状态/条件下蛋白质表达发生的显著变化。PEAKS® Q 定量模块支持LC-MS/MS 的各种标记或非标记定量方法,实现多样本间的相对蛋白质丰度定量。
功能特点
- 基于定性和特征峰的DDA定量
- 支持非标记和标记定量:LFQ,TMT(MS2/MS3),iTRAQ,SILAC等
- 支持复杂实验设计同时定量
- 详尽的数据和结果可视化展示
- 基于碎片离子的DIA定量
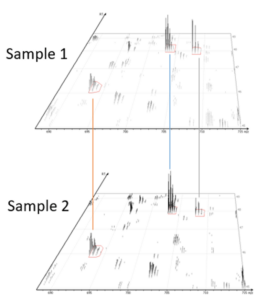
基于定性和特征峰向量的LFQ算法逻辑
基于定性的LFQ
- 特征峰检测
- 特征峰定性关联
- 保留时间对齐和特征峰匹配
- 相对丰度计算
- 显著性计算
基于特征峰向量的 Q
- 特征峰检测
- 保留时间对齐和特征峰匹配
- 相对丰度计算
- 显著性计算
- 特征峰定性关联
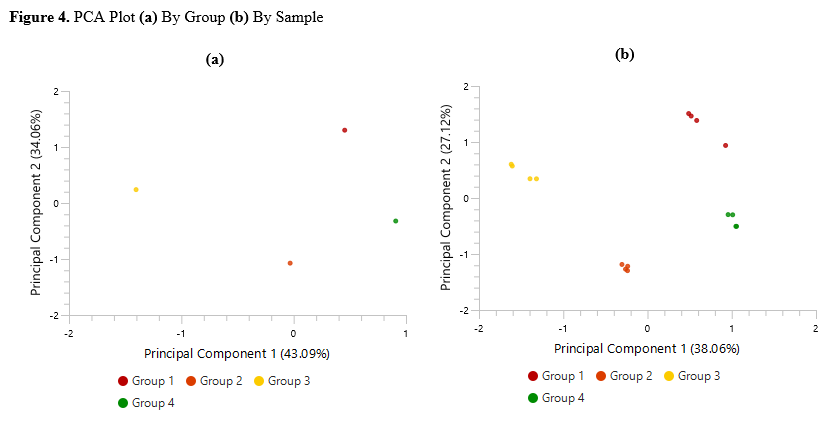
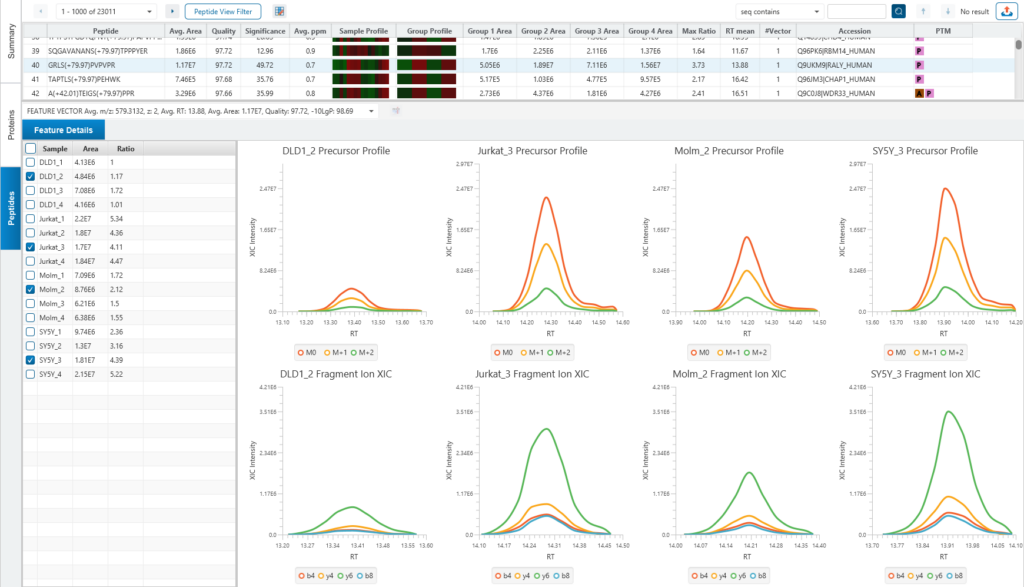
丰富的数据和结果可视化展示:热图、变化趋势图、火山图以及 PCA
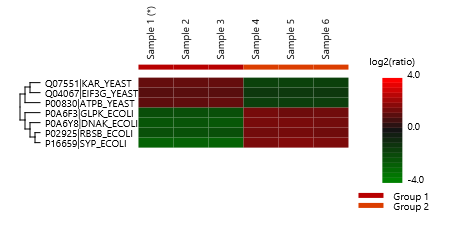
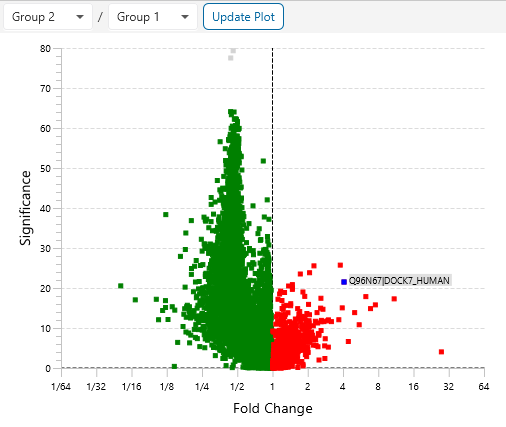
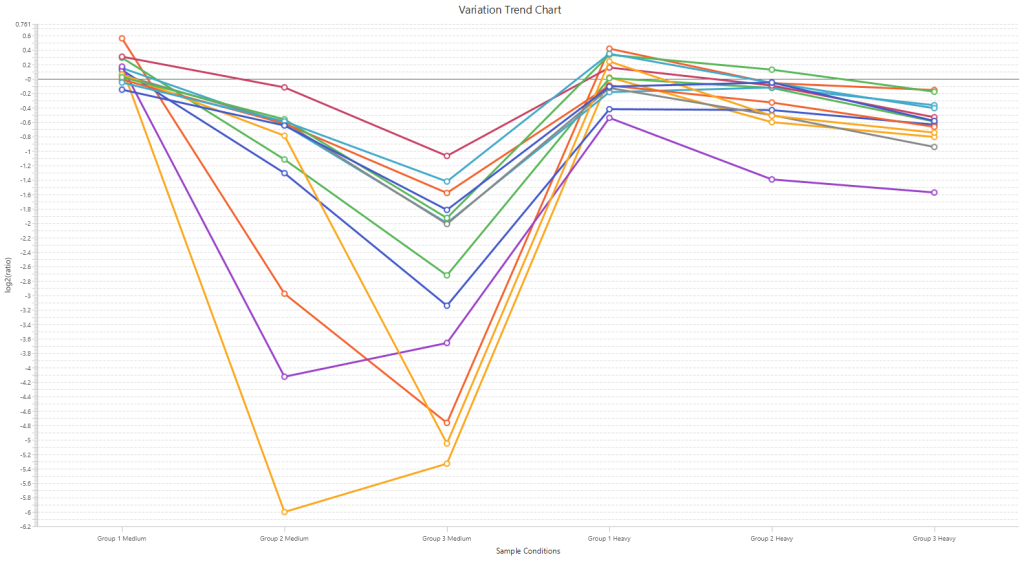
细胞培养过程中的氨基酸稳定同位素标记技术(SILAC) 是复杂蛋白样品相对定量的一种经典方法, 该技术现在已经成为了体内同位素标记最常用的蛋白质组学方法。
PEAKS® Q模块采用以下策略分析SILAC数据:
1.优化SILAC特征峰检测方法
2.SILAC轻重标特征峰匹配和ID-transfer提高定量的准确性和灵敏度。采用了比值形式进行定量,支持super-SILAC实验的数据分析。
通过引入多组比较,可以实现时间梯度的定量。PEAKS® Q模块能够实现分别针对单组分析和多组比较分析的配对t检验和ANOVO统计分析。点击了解更多信息
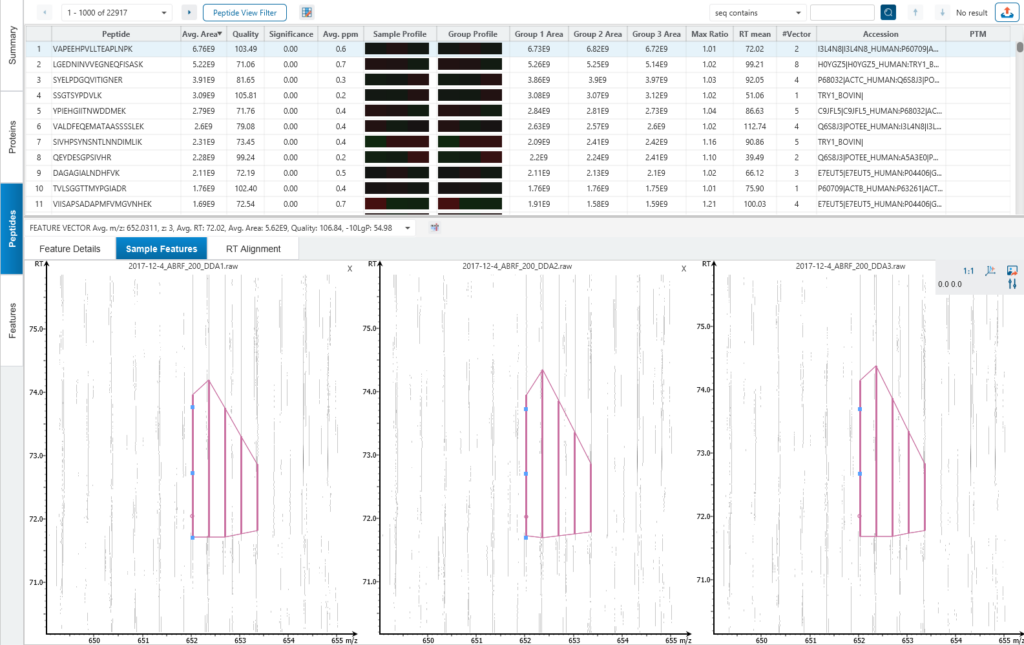
PEAKS® LFQ通过MS1和MS2离子峰强度分别对DDA和DIA数据进行定量。对于LFQ的实验,样本一般是单独收集、处理并采集质谱数据,因此当样本较多时会产生大规模数据,PEAKS®通过高灵敏算法自动进行离子峰比较和匹配,实现准确定量。
对于DDA数据,PEAKS®LFQ基于MS1特征峰计算相对丰度。默认设置下,使用可被定量的前三名unique peptides计算相应蛋白的表达值,修饰肽段默认排除,也可手动配置。
对于独立或者相关样品进行分析比较时需要采用不同的t检验或ANOVA方法。在进行较大数量的检验分析时,对于更准确地估计错误发现率(FDR),P值的解释也会有所不同。了解更多
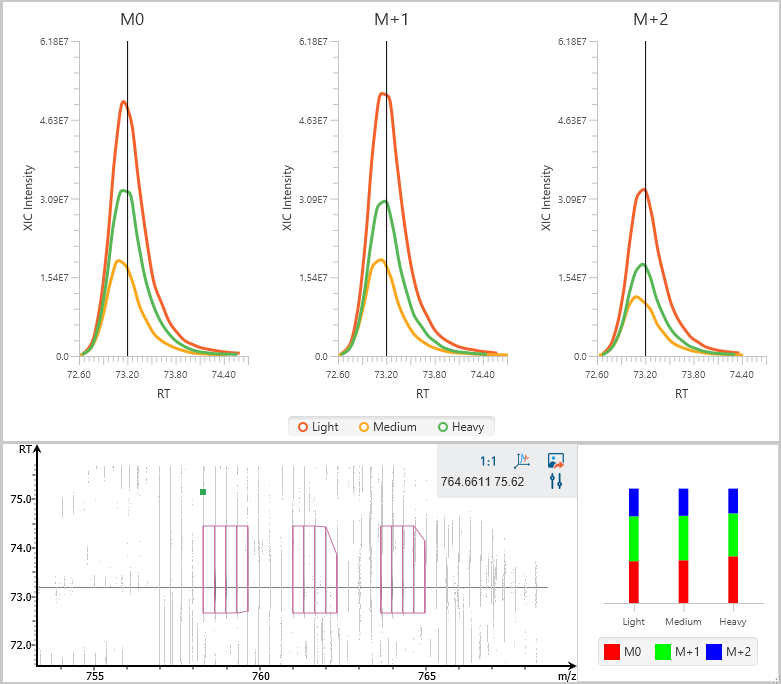
PEAKS® 的DIA蛋白相对定量是基于MS2碎片离子峰强度计算的。PEAKS® 12通过AI驱动的先进算法自动选择合适的离子,基本的选择依据包括:
1.所有样本中都有峰
2.色谱流出连续
3.与深度学习预测的保留时间和二级谱匹配
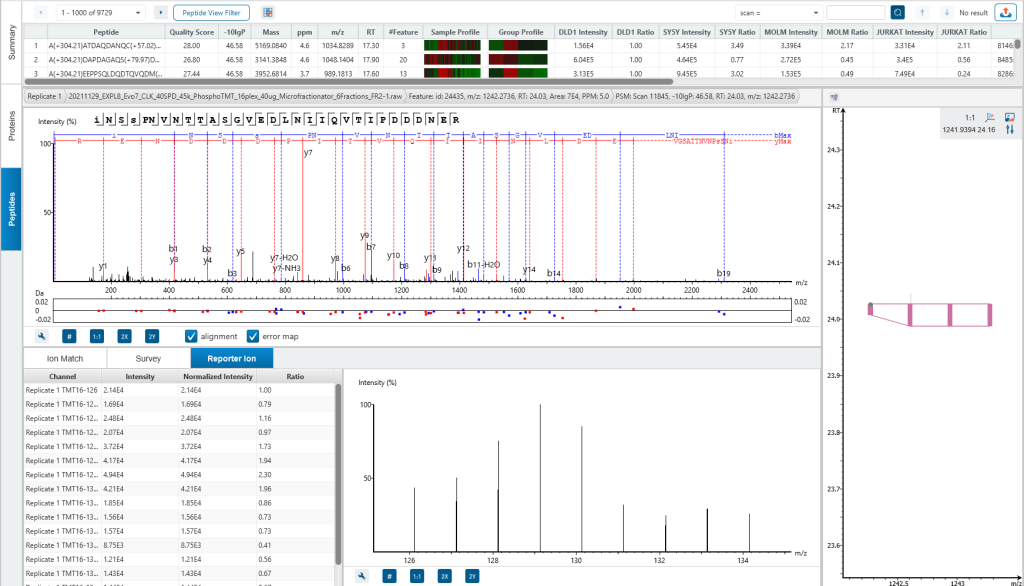
同位素标签 (TMT或iTRAQ)具有相同的质量和化学性质,使得轻重同位素可以共洗脱。这些标签在MS/MS中通过碰撞诱导离解(CID)被从肽段上分离下来,进而用于定量分析。
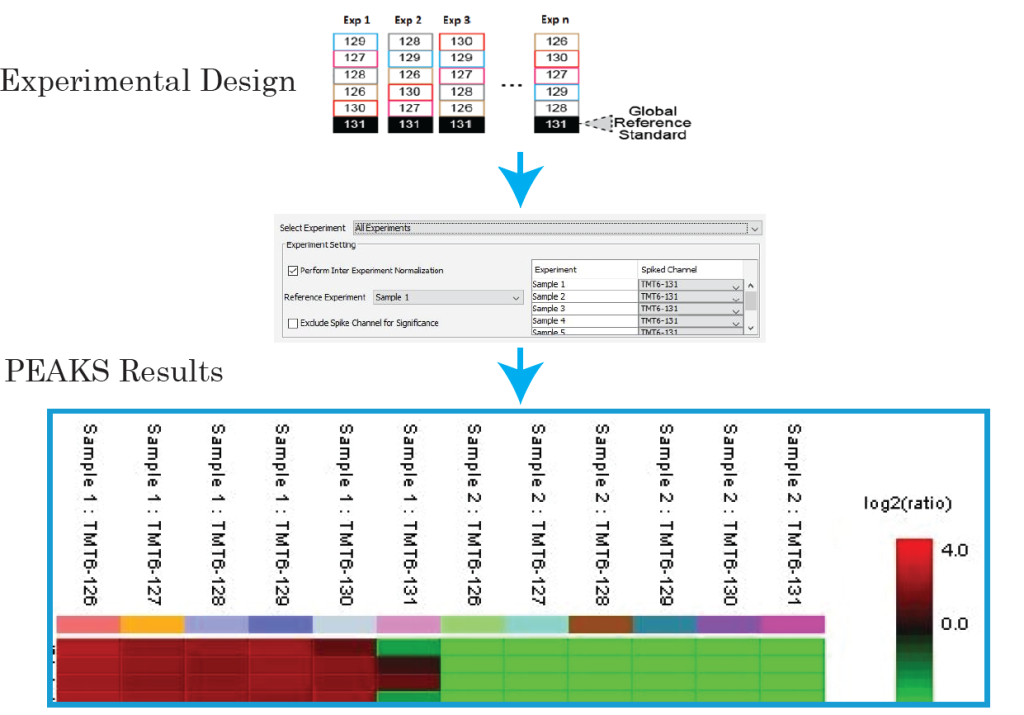
这个方法的局限之一是在质谱鉴定的过程中TMT/iTRAQ标签的报告基团含量的测定可能会受到其他碎片离子的干扰。MultiNotch MS3方法通过将多路复用技术和定量灵敏度及精确度结合在一起解决了这个问题。PEAKS®同时支持MS2和MS3定量方法。
有了PEAKS®,您现在可以扩大样本数量进行大规模的蛋白质定量研究,需要内参标签以确保多组标记之间的定量准确性。 了解更多
References
- Yang, W., et al. PEAKS Q: Software for MS-based quantification of stable isotope labeled peptides. ASMS. WP531. 30/5/2006.
- Xin, L. et al. New Quantitation Software Package Based on PEAKS Protein ID. ASMS. TP 653. 2/6/2008.
- Chen, C., et al. New Algorithm for Label-Free Protein Quantification. ASMS. MPB 043. 31/05/2009.
Resources