
 SPIDER
SPIDER
序列突变 & 同源搜索
生物样品中的蛋白质序列通常和数据库中相应的蛋白序列有略微差异,这些差异很多是由蛋白多态性、抗体多样性、数据库误差、跨物种数据库搜索等造成的。序列突变信息的缺失则很可能漏掉潜在的biomarker、抗体确认时引入错误、甚至蛋白质会有较低的覆盖率。对于这些问题,PEAKS SPIDER模块设计用于检测序列的突变信息并进行跨物种的同源搜索。
概览
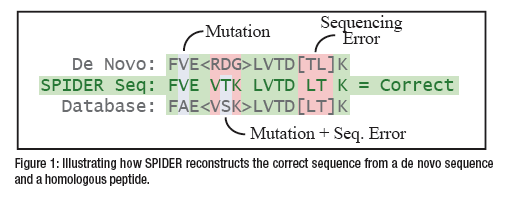
PEAKS SPIDER算法将de novo sequence tag和database protein进行匹配,发现序列之间存在显著性的相似,算法则通过从头测序错误和同源肽段序列突变来解释这个差异(Figure1)。更具体说是重建一个”real sequence”,使得正确序列和de novo结果之间的从头测序错误、正确序列和数据库蛋白序列的同源多肽突变,这二者的总和最小化。
为什么不用BLAST?
常规的同源搜索工具比如BLAST并不是搜索de novo sequence tags的好的选择。由于肽段的MS/MS谱中缺失一些片段离子导致可能发生从头测序错误的现象是很常见的。因此,对于合适的de novo tag的同源搜索,要有如(AT/TA)和 (N/GG) 这样的测序容错。而BLAST出于不同的考虑,会拒绝掉错误太多的情况,可能显著降低了搜索灵敏度。另外,BLAST也不会进行真实肽段序列的重建。
除了明显的突变检测和跨物种搜索功能外,SPIDER另外一个非常有用的应用是可以迭代地使用它来进行一个完整蛋白质的测序(如抗体测序),通过以下步骤实现:
- 使用PEAKS标准workflow (de novo + PEAKS DB + PEAKS PTM + SPIDER)进行同源数据库搜索,可以鉴定同源蛋白质。
- 然后再覆盖窗口中选择工具“copy mutated protein sequence”,这样可以把突变的蛋白质序列(after applying the confident mutations)复制到Windows剪贴板上。
- 通过粘贴复制的序列作为蛋白质数据库调用另一个标准搜索。
- 多次重复以上步骤,逐步提高序列质量。
发现具有氨基酸水平可信度的突变
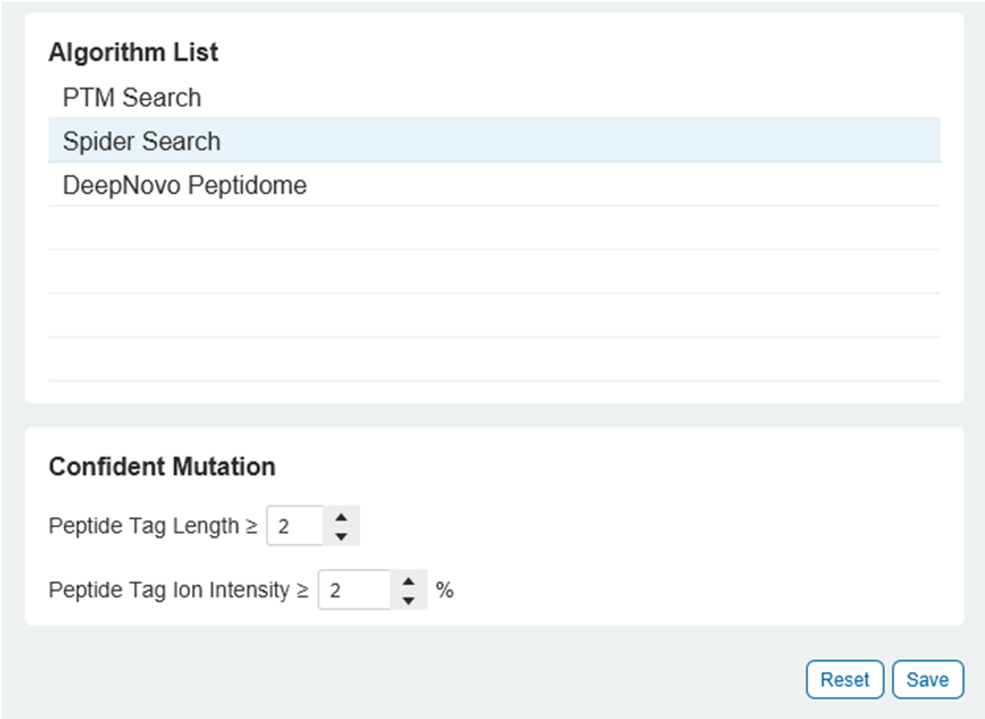
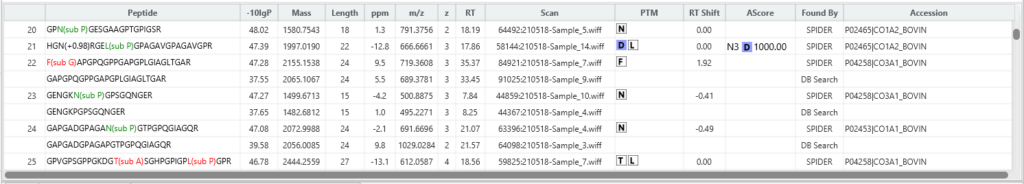
新的PEAKS 12 Spider分析提供了下一级的置信度,并使用户能够设置肽标签长度和肽标签离子强度阈值,超过该阈值,突变被认为是可信的。肽标签长度是突变位点上需要具有高于突变设定值的离子强度才能被认为是可信的连续离子的数量。如果突变发生在N末端或C末端,那么需要至少有一个额外的连续离子比所选的突变数量多,才能被认为是可信的。SPIDER结果现在包含一个新的Mutated Peptide tab,用户可以在其中看到哪些突变超过了他们设定的置信阈值。
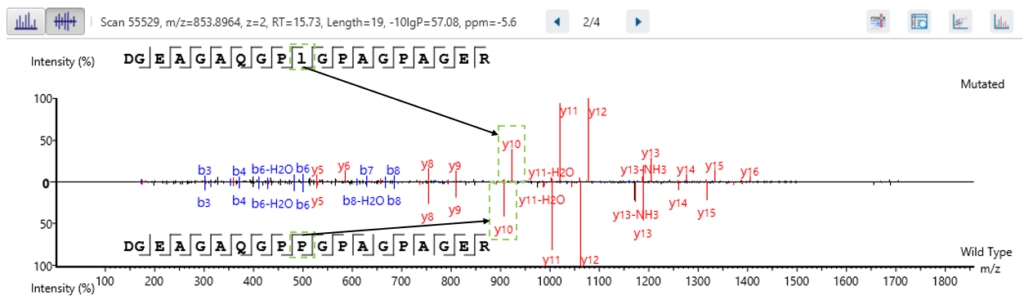
如果在样品中同时鉴定出原始和突变肽,则可以选择将MS2谱作为Mutated Peptide tab中的镜像图来查看。这可以方便地查看支持离子。
- Han, Y., Ma, B., Zhang, K. SPIDER: Software for Protein Identification from Sequence Tags Containing De Novo Sequencing Error. Journal of Bioinformatics and Computational Biology. 3(3):697-716. 1/6/2005.
- Ma, B. & Johnson, R. De novo Sequencing and Homology Searching. Molecular & Cellular Proteomics. 11(2). 1/2/2012.