
PEAKS DB 功能特点
- 基于LC-MS/MS 数据的搜索引擎
- 未知翻译后修饰搜索及分析
- 序列突变搜索
- 与从头测序整合
- 支持CID, HCD, ETD/ECD, EThcD、EAD等多种碎裂模式
蛋白的质谱鉴定
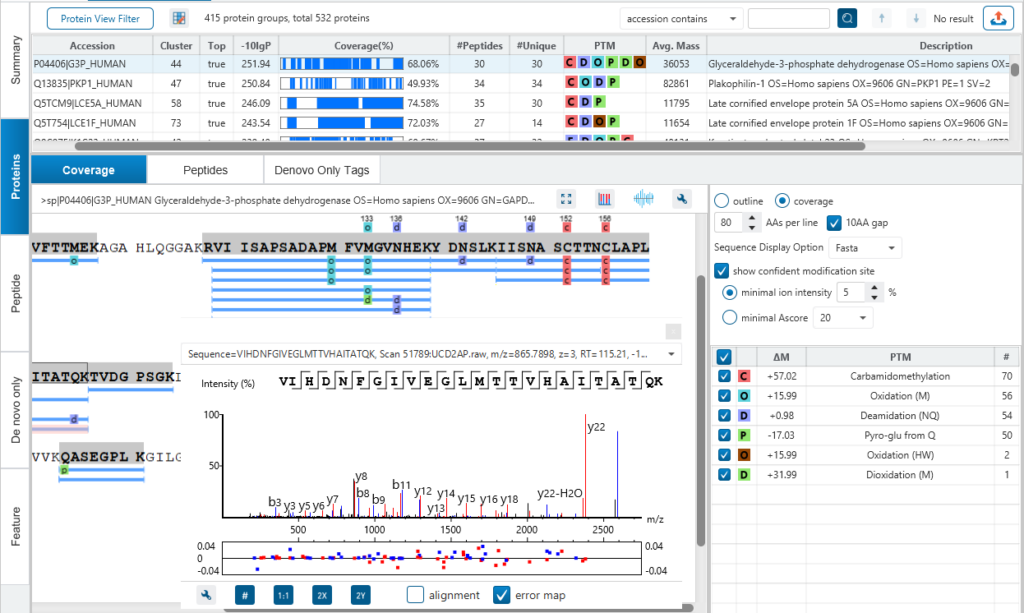
自下而上的蛋白质组学(LC-MS/MS)是当今许多蛋白质研究的基础。对于肽段碎片数据的解析有多种算法。一般的算法通过比较实验检测到的碎裂离子列表和从蛋白质序列数据库中获得的理论碎裂,基于概率的匹配来鉴定肽段。PEAKS DB在进行蛋白鉴定时集成了数据库搜索和从头测序。因此,它提供了完整的多肽鉴定,包括各种修饰,突变和未报道过的多肽。
灵敏度和准确度
PEAKS DB是基于sequence tag的搜库算法,保证了鉴定的准确性和灵敏度。独特的“decoy-fusion”为PEAKS DB提供了非常准确的错误发现率(FDR)评估方法。这说明PEAKS DB是一种更加准确、更加灵敏的蛋白鉴定搜索引擎。
各种数据库搜索引擎在多肽鉴定上的性能
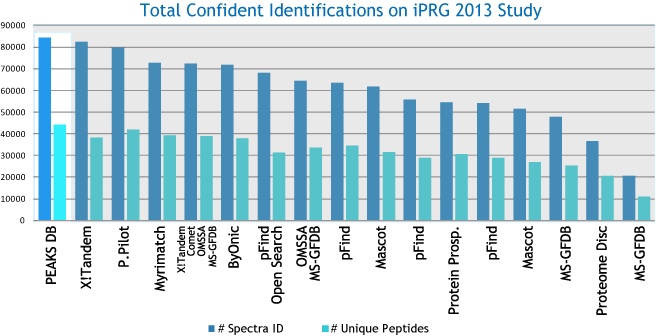
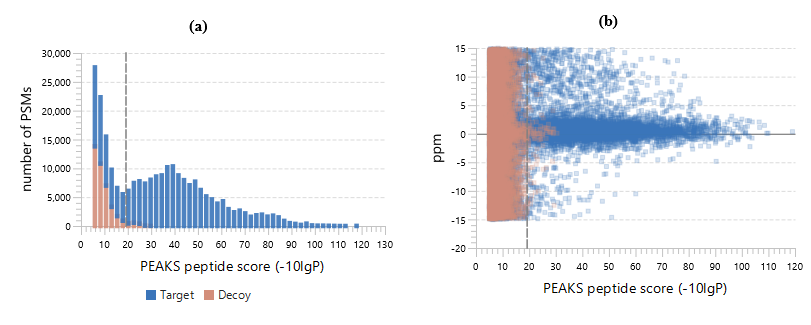
PEAKS DB可以自动地对蛋白鉴定数据库搜索任务进行统计分析,并在Summary Page中显示统计数据。用户可以通过指定的FDR轻松地过滤数据库搜索结果。
结果可视化
PEAKS 一直以其卓越的结果可视化而闻名。除了以上所述的总览视图,用户还可以从不同角度方便地查看蛋白质鉴定结果。
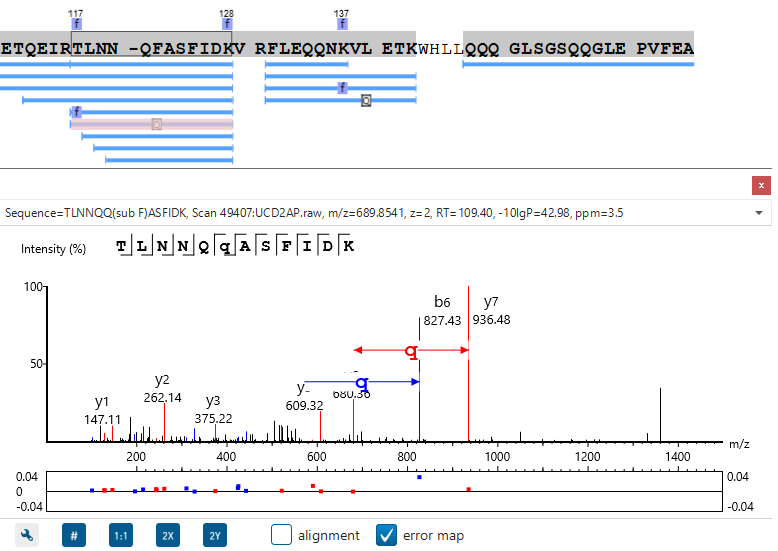
特别是,蛋白质覆盖视图(Protein Coverage View)为用户提供了非常有用的信息,包括在一个蛋白中鉴定到的所有多肽,同时多肽的修饰位点信息和序列突变均会以高亮的字符显示。
当用户点击其中一条鉴定的多肽,PEAKS可以提供肽谱匹配的注释信息,同时当鼠标在氨基酸残基上移动时,相应支持的碎片离子信息会在谱图实时显示。
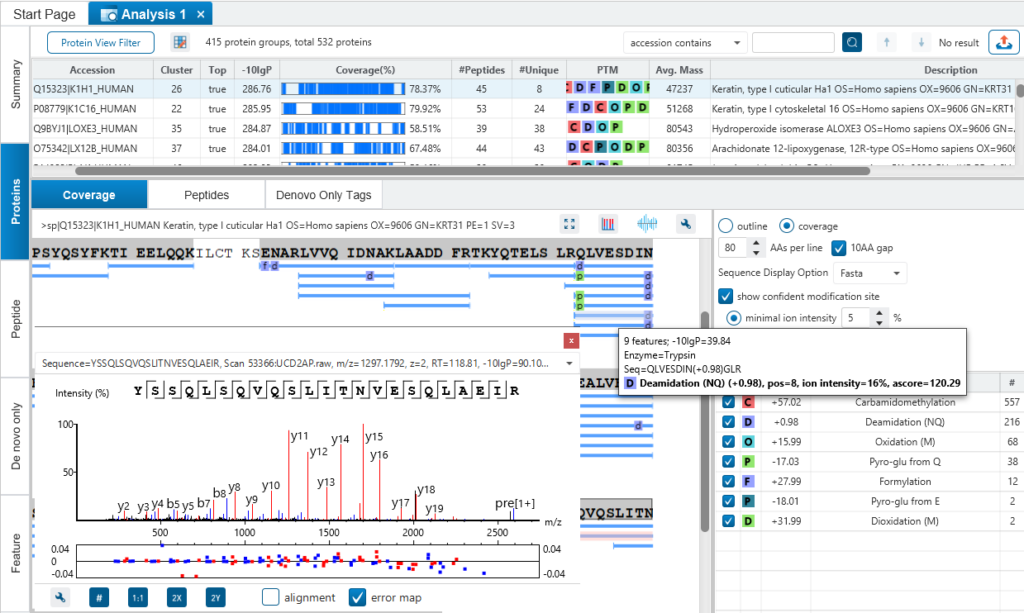
与 de novo Sequencing结果整合
同源多肽鉴定
传统的搜索引擎的假设是,数据库中包括了我们样品中的蛋白质的序列信息,但是,实际上很多我们关心的重要蛋白质,例如单抗,并不包含在任何常用数据库中。所以这种搜库策略的缺陷在于,搜库得到的蛋白最多可以尽可能匹配用于检索的数据库,但是当数据库本身不完整的时候,就会造成大量的多肽不能被鉴定或者鉴定错误。
整合了de novo sequencing 的PEAKS DB,能够提供更为完整的肽段的鉴定,而不依赖于多肽是否存在于数据库中。
Reference
Tran NH, Qiao R, Xin L, Chen X, Liu C, Zhang X, Shan B, Ghodsi A, Li M. Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nature Methods. 16(1), 63-66. 20/12/2018.