
PEAKS软件概览
基于质谱的蛋白质组学定性定量分析软件解决方案
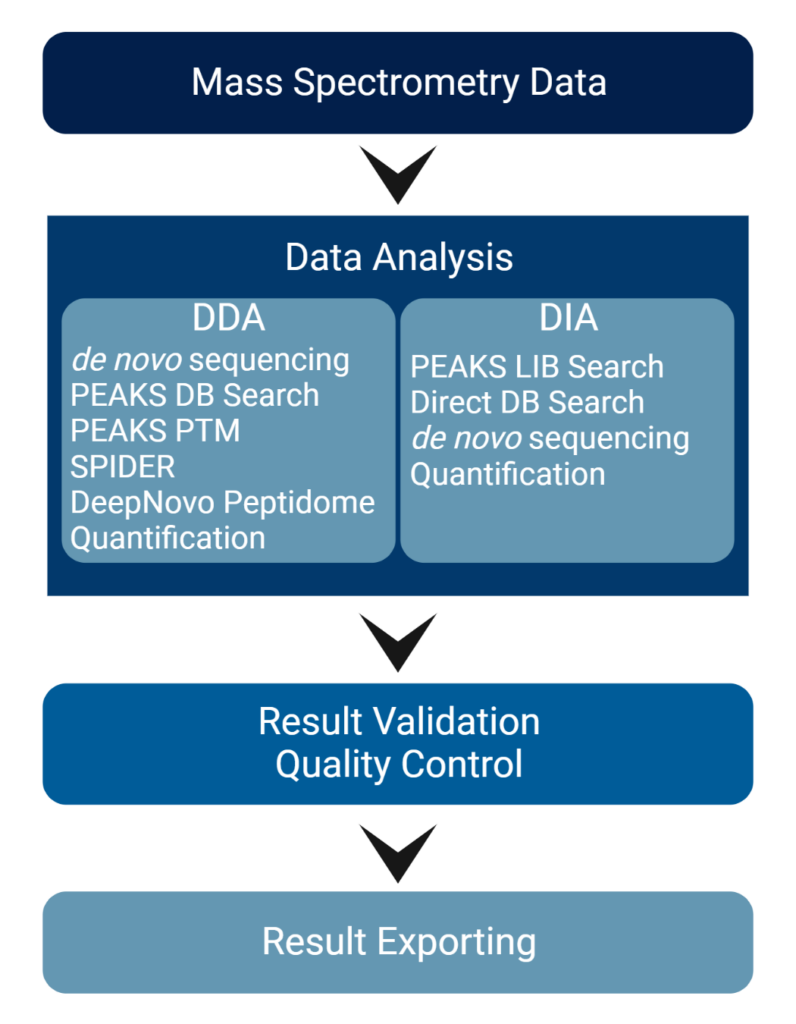
PEAKS系列软件产品是针对基于液相质谱方法的蛋白质组学数据的完整解决方案,对复杂的蛋白质样品中的多肽或蛋白质进行定性和定量的系统性分析。PEAK软件是仪器厂商中立的分析平台,可以直接从质谱仪加载原始LC-MS/MS数据(无需提前转换数据),在PEAKS工作流程中选择进行肽和蛋白质鉴定(如从头测序,数据库和谱库搜索),翻译后修饰(PTM)分析、突变的表征以及定量分析。PEAKS还为数据可视化、结果验证和报告提供了详细、易于使用的用户交互界面。
与其他仅依赖数据库搜索的软件工具不同,PEAKS使用独特的从头测序辅助序列数据库搜索算法,最大限度地提高肽段的鉴定效率,对复杂蛋白质组提供了更深度的解析。PEAKS 基于深度学习的技术,整合了多种算法包括从头测序(de novo sequencing),数据库搜索和谱图库搜索,集成在用户易用的工作流中,通过使用这些方法,您可以期望提高蛋白组/多肽组质谱数据的准确性和灵敏度,赋能未知修饰和突变体的发现,并从从头测序的肽段结果中推断新的ORF。
PEAKS软件提供多种部署方案,以适应各种计算环境,如桌面工作站、局域网内搭建的服务器和云环境。

PEAKS Studio
桌面版解决方案

PEAKS Online
服务器高通量解决方案
产品优势
PEAKS直接支持来自各大主流质谱仪器厂商的所有原始文件格式,无需任何转换。使用原始数据的优势在于,通过将这些原始数据的底层数据库嵌入到PEAKS算法中,数据中呈现的信息可以达到最大化。无论您在实验室中使用哪种仪器,PEAKS都针对不同类型的仪器进行了相应的算法训练,旨在确保您分析结果的最佳准确性和灵敏度。
DDA和DIA技术都在迅速发展,研究人员需要一种结合两种采集方法优点的分析方法。在工业界,DDA仍然是常用的方法,这种方法相对已经完善,对于尚未被很好地表征的样品尤其重要。近年来,DIA越来越受欢迎,因为它具有在选定的m/z范围内并行地获取所有的母离子的所有片段离子的特性。这克服了DDA中连续MS/MS采集的局限性。
全新DIA工作流
PEAKS®️为DIA数据分析提供稳健的解决方案。在多肽的鉴定中,结合了三种数据分析方法:谱图库搜索,直接序列数据库搜索(direct database search),和从头测序。这样的搜索方法,其搜索空间逐渐放大。首先,可以通过由先前的谱图鉴定结果产生的谱图库进行谱图库搜索,通过了FDR设定阈值过滤后的多肽被保留下来,没有通过谱图库搜索FDR阈值的可以继续进行直接序列数据库搜索。其中可信的数据库匹配结果添加到结果中。然后使用同样的FDR方法,在数据库搜索中未匹配的谱图进一步执行从头测序。
PEAKS 11 为用户提供谱图库搜索的鉴定工作流。通过深度鉴定的谱图库,用户可以轻松快速地评估他们的数据。
谱图库查看器
在PEAKS 11中,用户可以通过PEAKS谱图库查看器来直观地了解他们谱图库的细节,便于通过数据的统计信息来评估谱图库的质量。这使得科学家可以轻松地自定义、检查和验证谱图库,以提高鉴定。通过直观的用户界面,允许您上传和查看由PEAKS Studio和PEAKS Online生成的文本格式PEAKS谱图库。
注意:该功能仅在PEAKS Studio中支持,PEAKS Online的用户可以免费使用PEAKS Studio的view模式来查看。
PEAKS以其卓越的结果可视化而闻名。用户在结果中可以访问几个不同的视图,例如,蛋白质视图,多肽视图,甚至在氨基酸水平检查。在PEAKS的蛋白质覆盖视图中,用户可以找到感兴趣的肽,并调出相关的谱图。即使是PEAKS的初学者也可以通过友好的工作流设置轻松地分析数据,内置的结果验证可以确保结果质量并为用户提供更多可信度的信息。PEAKS图形用户界面设计主要从易于解释,结果可视化和验证的角度来设计的,以帮助研究人员从中获取分析项目的全局信息。
PEAKS通过将从头测序得到的多肽序列与相应的数据库搜索的肽谱匹配结果相结合,为其数据库搜索工作流提供了一种独特的方法。从头测序的多肽序列与蛋白质数据库条目进行比对,能够为用户提供关于PTMs、突变、同源肽和全新肽段的额外信息。PEAKS DB使用基于序列标签的搜索算法,保证了搜索的准确性和灵敏度。使用PEAKS DB,研究人员可以通过分析数据库中或不在数据库中的肽来深度表征他们的样品。了解更多
在PEAKS DDA工作流程中,通过深度学习技术大大提高了性能,最大限度地提高PEAKS DB搜索结果中的多肽鉴定效率。采用深度学习的算法来预测保留时间、碎片离子强度、质荷比和离子迁移率,提高了鉴定的准确性和灵敏度。
通过整合PEAKS DB和de novo测序结果实现对PTM的深度鉴定。PEAKS领先的算法最大限度地提高了PTM鉴定和PTM profiling。常规的PTM可以在任意的定性分析(从头测序、PEAKS DB、谱库搜索、PEAKS PTM和SPIDER)中定义,然而,只有在PEAKS PTM搜索中,用户才能通过指定一组感兴趣的PTM列表或从Unimod数据库中搜索所有313种天然发生的生物修饰来扩展PTM分析,以发现数据中任何预期以外的PTM。PEAKS PTM通过集成强大的从头测序算法和数据库搜索,专门用于发现潜在发生的修饰,并且不会过于受到计算资源的限制。使用PEAKS可以最大限度地提高鉴定效率,并彻底表征复杂蛋白质组中的翻译后修饰。
PEAKS软件中的SPIDER,是一种专门用于检测多肽突变并进行跨物种同源性搜索的算法。SPIDER算法尝试将de novo序列标签与数据库蛋白质匹配。当发现显著的相似性时,算法尝试使用从头测序错误和同源肽突变来解释差异。SPIDER旨在重建一个“真实”序列,以最小化真实序列与de novo序列之间的从头误差之和,以及真实序列与其之间的同源肽突变。可以通过PEAKS以从头测序为基础的同源搜索,来获得数据库中以外的可靠匹配。了解更多
这是一个专门为多肽组学数据分析的应用场景而开发的全新专属工作流,结合了数据库搜索、从头测序和多肽突变的鉴定。利用多肽组学数据集训练DeepNovo深度学习模型,可显著提高多肽鉴定的灵敏度和准确性。此外,将de novo peptide (non – canonical)与数据库peptide (canonical)相结合,在全局水平更准确地估计FDR。最终输出结果中的多肽被分类为来自于数据库匹配,DeepNovo或同源多肽(突变肽),可以直接输出用于结合亲和力和免疫原性预测。了解更多
注意:本工作流目前应用在PEAKS Studio和PEAKS Online产品中
为了了解复杂生物系统中单个蛋白质的功能,通常需要测定蛋白质丰度的变化。作为一个可选的附加模块,用户可以使用PEAKS Q,通过LC-MS/MS标记或非标记定量的方法同时确定一组样品的相对蛋白质丰度变化。了解更多
注意:本工作流目前应用在PEAKS Studio和PEAKS Online产品中
离子淌度质谱(IMS- MS)通过增加离子分离的第四个维度,为复杂的生物和化学混合物提供了令人瞩目的分析流程。离子通过缓冲气时,根据其迁移率进行分离,使用IMS-MS,可根据其大小、形状、电荷和质量迁移率区分离子。因此,在传统质谱中可能无法区分的离子,有可能通过这种方式区分开来。
PEAKS IMS模块,整合在PEAKS任意工作流中。易于使用的PEAKS图形用户界面将原始数据分为IM-MS, IM-MS/MS和LC-IM/MS。研究人员可以很容易地查看基于离子迁移率的四维特征谱峰。了解更多
PEAKS Glycan是PEAKS Studio中的可选模块,为深度糖蛋白组的分析提供了独特的解决方案。PEAKS Glycan利用基于完整糖肽的方法,针对LC-MS/MS数据分析样品中的糖蛋白。PEAKS Glycan模块提供了一个完善的数据库搜索和新开发的算法,以促进N-和o -链聚糖的鉴定和表征。Glycan Profiling是在蛋白质的特定位点进行的(位置分析),并通过非标记定量比较样品中的糖肽丰度。
如何选择组学解决方案
| Features | PEAKS Studio | PEAKS Online |
|---|---|---|
| 数据类型 | ||
| CID/CAD/HCD/ETD/ECD/EThCD | √ | √ |
| Mixed | √ | |
| DDA | √ | √ |
| DIA | √ | √ |
| IMS/MS | √ | √ |
| 工作流/算法 | ||
| 最新PEAKS 算法版本 | 11 | 11 |
| Manual De Novo | √ | |
| Auto De Novo | √ | √ |
| Database | √ | √ |
| DIA workflow (Spectral Library Search + directDB + de novo sequencing) | √ | √ |
| PEAKS PTM | √ | √ |
| SPIDER (突变分析) | √ | √ |
| PEAKS DeepNovo Peptidome | √ | √ |
| Label-Free Quantification | √ | √ |
| Label Quantification | √ | √ |
| 工作流管理与数据组织 | ||
| 保存参数与工作流,建立标准化分析流程 | √ | √ |
| 用户角色与安全管理 | √ | |
| 支持自动化工作流执行:允许执行多步数据分析流程,无需用户在每一步骤中进行设置 | √ | |
| 序列数据库管理(添加/删除/配置) | √ | √ |
| 数据结果报告与导出 | ||
| 数据/结果导出文本文件 (.csv, .fasta, .tsv) | √ | √ |
| 数据/结果导出标准数据格式文件 (.mzML, .mzXML, .mgf, .pepXML) | √ | √ |
| Html格式结果导出,包含可视化信息 | √ | |
| 产生谱图库 | √ | √ |
| 可以使用“查看模式”应用程序查看结果 | √ | √ |
| 技术需求 | ||
| 支持多用户 | √ | |
| PEAKS 分析项目的可备份与还原 | √ | √ |
| 支持Windows 10、11或server版 | √ | √ |
| 支持Linux系统 | √ | |
| 支持执行至少2个并发的数据分析工作流 | √ | |
| 可以部署到可弹性扩展的云环境,例如AWS | √ | |
| 高通量解决方案 | √ |