PEAKS IMS 附加模块
离子淌度质谱(Ion-Mobility Spectrometry Mass Spectrometry,IMS-MS)通过增加额外的离子淌度维度,为复杂生物化学混合物的分析提供了引人注目的工作流程。使用PEAKS IMS模块,可以最大限度的解决DDA数据应用中分析深度的有限性,以及DIA数据应用中多路复用的MS/MS谱图分析困难性的问题。
功能特点
- IMS-MS数据投射在“m/z到RT”或“m/z到1/k0”层面上的交互式数据可视化工具
- 4D检测到的feature的分析
- 解析重叠的母离子扫描和嵌合光谱
- 支持 4-D 标记和非标记蛋白质定量
- DIA PASEF
IMS-MS原始数据可以直接加载到PEAKS中,无需任何预转换步骤。加载到软件后,PEAKS IMS模块可以在四个维度上分析数据:- m / z,保留时间,强度和离子淌度。这些向量的组合使数据能够投射到 m/z-rt 和 m/z-1/k0 维度上。
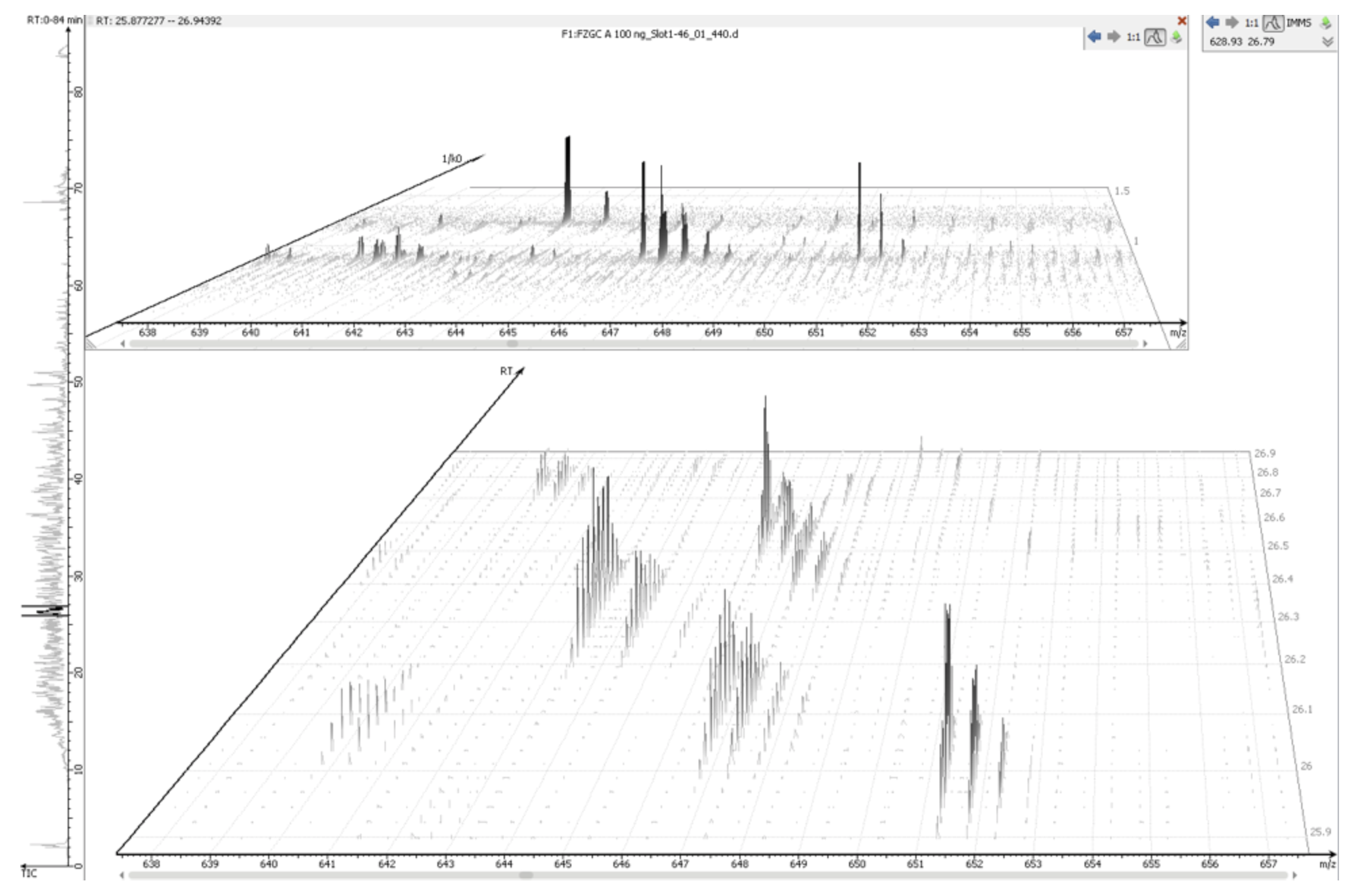
使用PEAKS IMS和从IMS-MS扫描中提取的4D信息,PEAKS可以准确鉴定和定量复杂生物样品中的肽,随后定量蛋白质,具有高度的准确度和灵敏性。
通过离子淌度分离,无论m/z和保留时间是否非常接近,都可以检测到两种不同的肽。而如果不提高灵敏度,重叠的前体会被忽略。通过利用IMS技术提供的所有维度信息,PEAKS IMS可以通过基于feature的方法和4D解卷积来解决发现蛋白质组学研究中常见的痛点,如磷酸肽位置异构体。下面的示例显示了共洗脱的同量异位素HEK磷酸肽,该肽只能在离子淌度维度上分离。
如果没有离子淌度分离,同量异位肽极难通过传统的蛋白质组学工作流程进行鉴定和定量。通过将IMS-MS数据与PEAKS IMS模块结合使用,克服了修饰位点的预测工作和定量工作的困难。
当与PEAKS Q模块配合使用时,PEAKS能够利用来自PEAKS IMS模块的4D信息,使用无标记或标记技术定量蛋白质和肽的种类。
除了在PEAKS Q中执行的保留时间对齐外,PEAKS IMS模块还允许PEAKS Q在离子淌度维度上进一步执行feature对齐。根据 MCP 中的 Meier 等人(2018)的说法,CCS 值具有高度可重复性,因此在此维度上的对齐可确保更准确的定量结果。PEAKS IMS使用4D特征对齐,可以将肽的可识别和可定量的动态范围最大化,用于分析生物样品。
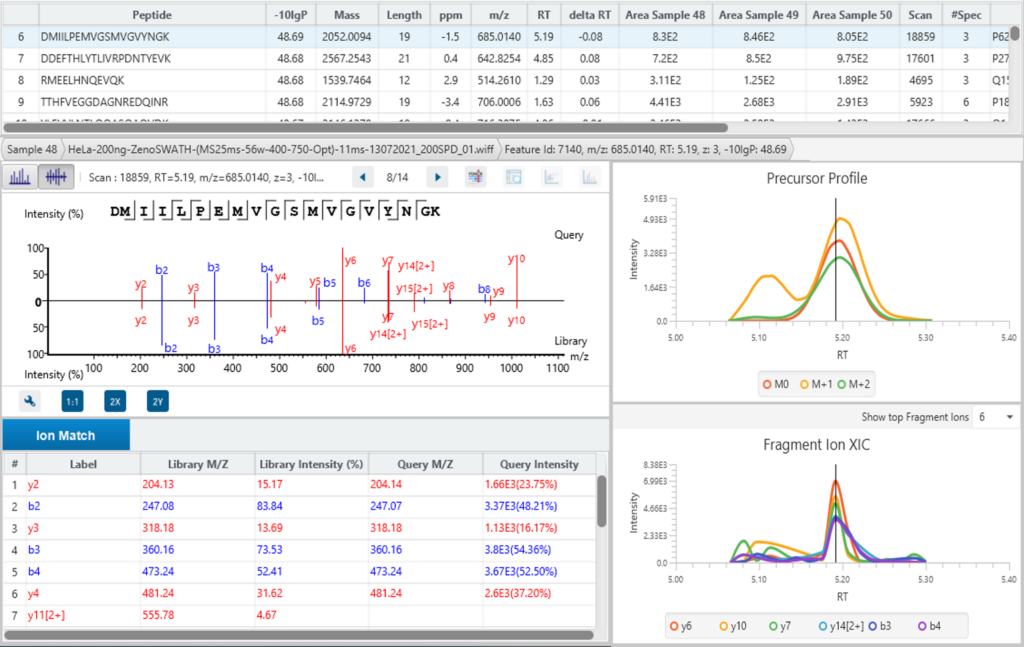
使用PEAKS IMS从数据依赖模式下获得的PASEF数据构建光谱库,并分析用Bruker的timsTOF Pro获得的diaPASEF(DIA)数据。
工作流程对肽母离子电流进行高达100% 的采样。这是数据非依赖性采集(DIA)的理想补充。DIA是一种并行数据采集方法,可在较大的母离子质量范围内捕获碎片离子。 Learn More
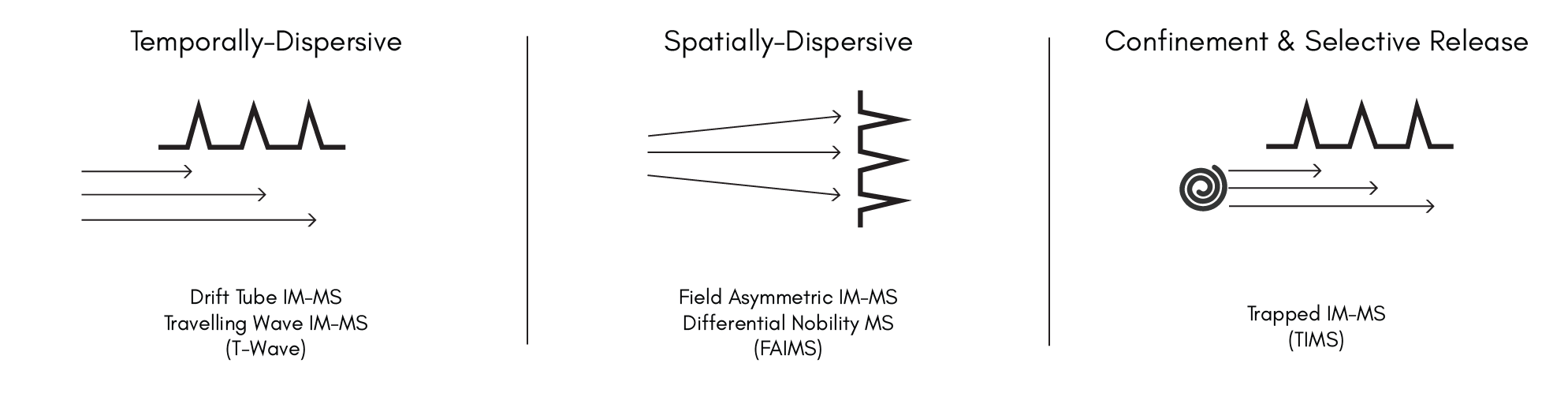
- May, J.C. and McLean, J.A. (2015). Ion Mobility-Mass Spectrometry: Time-Dispersive Instrumentation. Anal. Chem., 87 (3): 1422–1436.
- Meier, F., et. al. (2018). Online parallel accumulation – serial fragmentation (PASEF) with a novel trapped ion mobility mass spectrometer. Mol. Cell Proteomics, Dec 17 (12): 2534-2545.