

PEAKS GlycanFinder是一款用于深度糖蛋白质组学分析的综合性软件,可以很好地分辨糖链结构。它利用基于糖肽的方法来分析LC-MS/MS数据中的糖蛋白。PEAKS GlycanFinder集成了糖库搜索和糖的从头测序[1],提升N-和O-聚糖的鉴定和定量。并开发了基于深度学习的算法来解决糖基化位点和糖基结构的模糊匹配问题。
特色功能
- 从结构层面分辨N糖和O糖
- 结合多酶联合酶切方法,深度剖析糖基化位点
- 糖肽的非标记和标记定量分析
- 糖的从头测序帮助发现未知聚糖
- 结果图形可视化,便于快速验证
- 支持Orbitrap, timsTOF and ZenoTOF原始数据
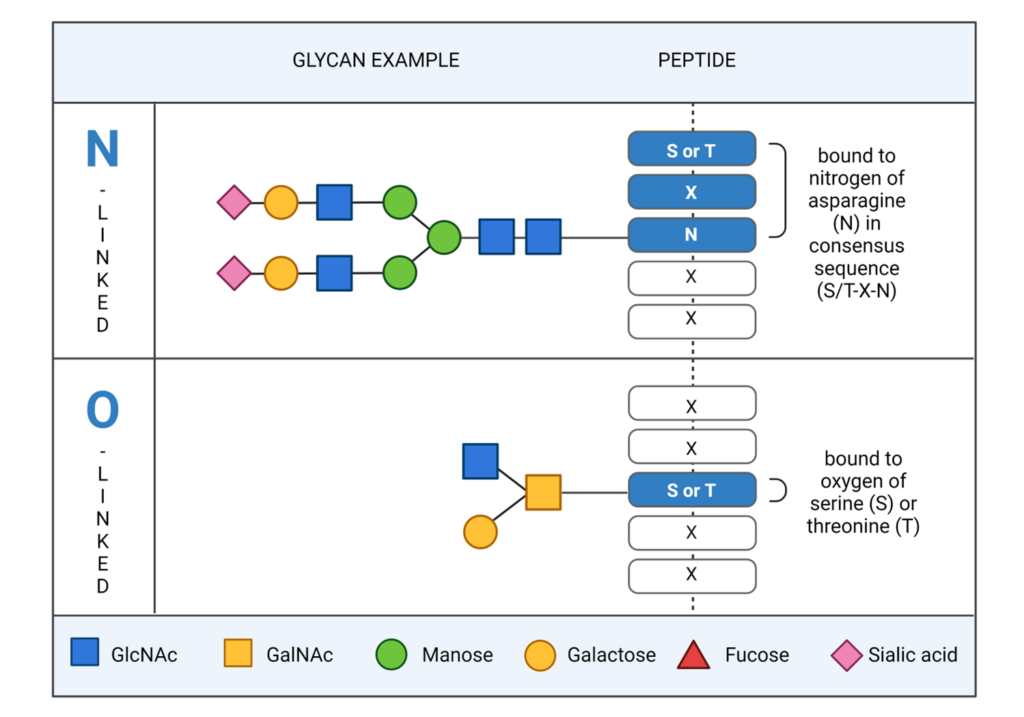
蛋白糖基化
蛋白质糖基化是指糖基或聚糖附着在蛋白特定氨基酸上的反应,是最重要的翻译后修饰之一,在众多生物学过程中起着关键作用。据估计,蛋白质组中50%以上的蛋白质是糖蛋白。聚糖是一种基于碳水化合物的异质结构,可以是线性的,也可以是支链的,附着在糖蛋白内的氨基酸侧链上。糖蛋白中的聚糖可分为N糖,即与天冬酰胺氮原子(N)结合,或O糖,即与丝氨酸(S)或苏氨酸(T)侧链的氧原子结合。重要的是,糖基化可以影响蛋白质的结构、稳定性和功能,对生理和病理功能都有很大影响。
糖蛋白质组学与质谱
糖蛋白质组学是从给定的细胞或组织蛋白质组中鉴定聚糖和相关的糖基化位点。来自MS/MS的碎片数据用于鉴定糖基化位点及其附着的N糖或O糖的结构。过去,基于LC-MS/ MS的糖蛋白质组学研究一直面临糖的结构异质性问题,即每个多肽可能存在多个糖基化位点,并且相同的聚糖组成可能包含多个不同的结构。
PEAKS GlycanFinder就是为了应对这些挑战所进行设计的。将所有肽谱匹配(PSMs)一致的结果用于确定糖基化位点。通过多种酶联合酶切,PEAKS GlycanFinder可提供非常准确的糖基化位点分析。如果多个候选糖链与某一糖谱相匹配,则为每个候选糖链计算一个S-Score。此外,糖的从头测序克服了糖库不完整的阻碍。
蛋白质视图
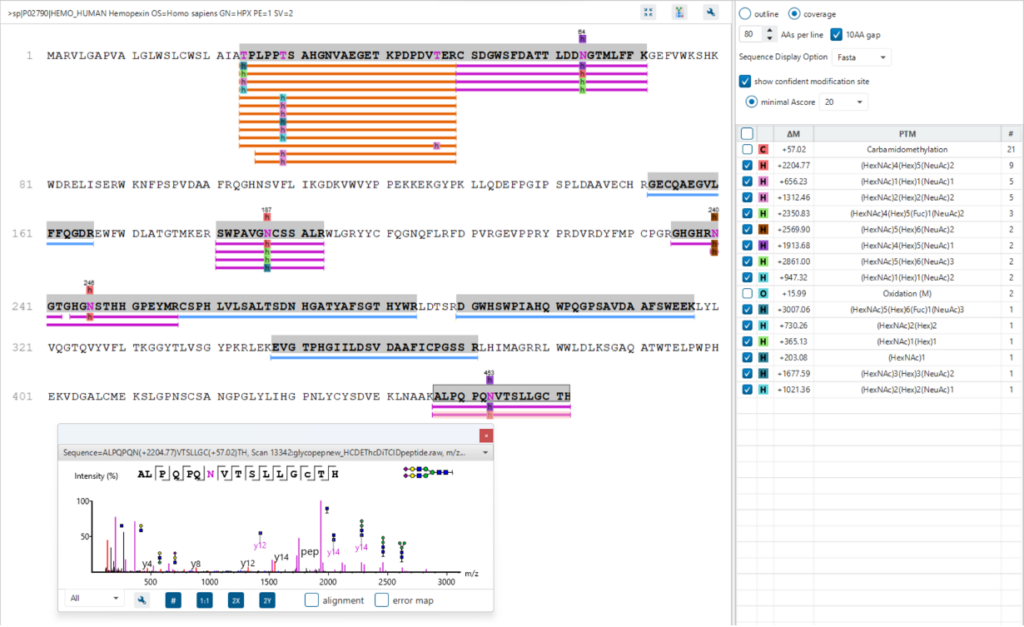
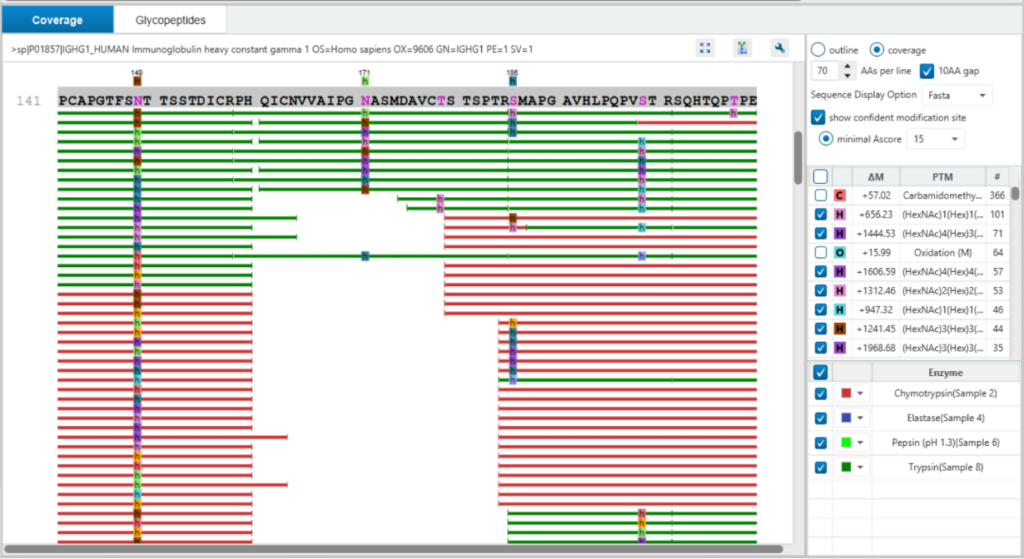
蛋白质视图(Protein View)显示的是从复杂生物样品中鉴定到的蛋白质。对于每个蛋白质,序列覆盖视图显示每个蛋白质的多肽匹配,并且结果是交互式的,因此用户可以直接点击查看每个肽谱匹配。用户可以很容易地通过颜色判断肽段类别,蓝色表示搜库匹配到的肽段,紫色表示N糖肽和橙色表示O糖肽。点击某一个蛋白质的糖基化位点(N,S,T为紫色),将以表格形式和饼状图的形式显示聚糖的分布。

多水平深度糖谱剖析
PEAKS GlycanFinder提供两种工作流用于分析糖蛋白质组学,即定性和定量工作流,包括糖基化位点分析(Glycan Site Profiling)和糖基化样品分析(Glycan Sample Profiling)。
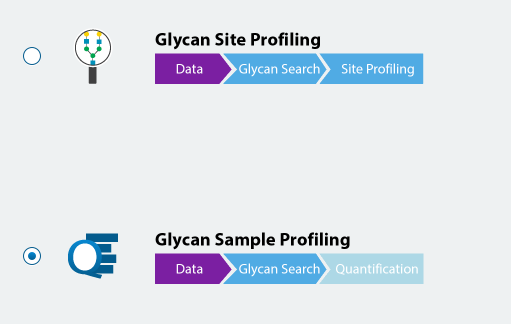
Site Profiling Workflow
多酶联合酶切揭示了每个糖基化位点的聚糖谱的细节。对于每个位点,1) 给出所有酶解多肽的糖基化位点一致性判断,2)在具体每一个糖基化位点给出所有酶中最佳的profiling结果。
Sample Profiling Workflow
通过Sample Profiling Workflow(样本分析工作流)可以获取蛋白的位点信息以及不同样本中糖肽的非标记和标记定量丰度。

价态匹配(Charge Lookup)
肽段经雾化电离后,通常会产生几种不同价态的母离子,但并不是所有母离子都能产生对应的二级谱。通常,PEAKS只计算有鉴定信息的母离子的特征峰面积用作该肽段的定量。因此,如果糖肽中某些价态的母离子未得到鉴定结果,该肽段的定量就会受到影响。为了得到更准确的糖肽定量信息,PEAKS GlycanFinder采用价态匹配(Charge Lookup)的方式来寻找同一多肽的所有母离子,然后将所有母离子的特征峰用于对应糖肽的定量。示例见下方图6,可以看到被 (HexNAc)4(Hex)5(Fuc)1(NeuGc)1修饰的EEQFN(+2075.73)STFR糖肽只有4个特征峰被鉴定到,但是在Glycan Chart中,用作糖肽面积计算的#Feature有5个,这就是通过Charge Lookup实现的。

糖肽视图
在PEAKS GlycanFinder中,糖肽视图(Glycopeptide View)提供两种糖基化肽段的谱图查看方式,镜像谱图注释便于验证糖链结构。

糖基化位点精确度(A-Score)
PEAKS GlycanFinder通过A-score在MS/MS水平对糖基化位点进行精确鉴定。A-score通过-10*log10(P)计算,P表示糖基化位点随机匹配的可能性。因此,A-score越大说明这条肽段上匹配到该糖基化位点的可信度越高。
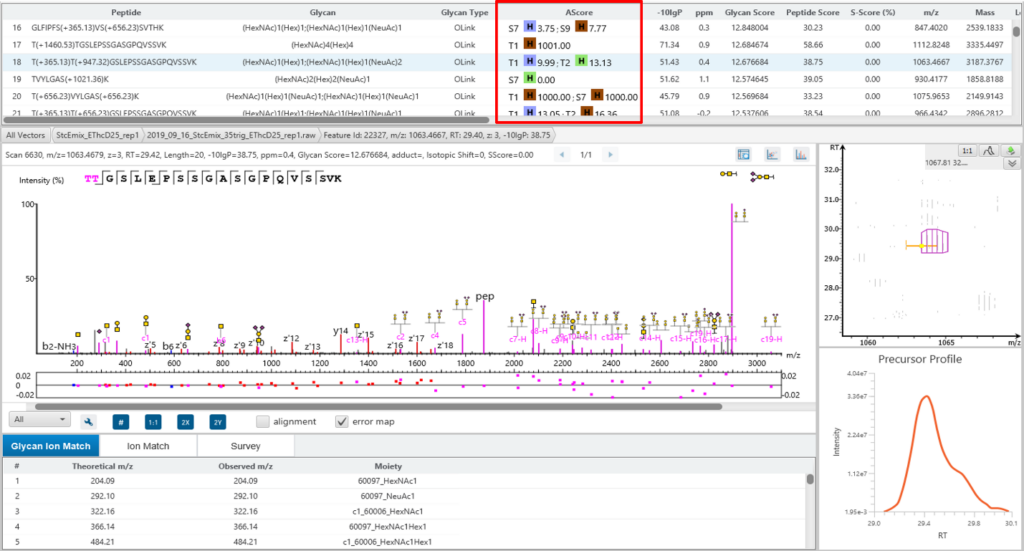
糖链结构特异性 (S-Score)
糖肽上的糖链都会给出一个S-Score(%),表示糖库中匹配到该糖链组成的糖型的匹配可信度。对于糖库中相同组成成分的糖型,候选糖链会按照糖的Y-ion数量排序,S-Score的计算公式为:S-Score = (most Y-ion count – 2nd most Y-ion count)/(most Y-ion count)。S-Score得分越高越好,100%意味着只有1个候选糖链结构并且匹配地非常好。0%意味着top1和top2的结构非常相似,无法确切地说哪个是最佳匹配。

糖的从头测序
当糖库中包含的糖不完全,或者肽段上发生了非预期的修饰时,PEAKS GlycanFinder的从头测序算法可以提供更多的糖肽鉴定结果。

糖库编辑工具
在PEAKS GlycanFinder中,用户可以通过糖库编辑工具自定义糖库,使用画板编辑和修改糖库中的聚糖并轻松导入软件。软件也可以同时提供糖的理论碎片离子信息。

糖蛋白质组学的LFQ和标记定量
在糖蛋白质组学的研究中,得到糖基化位点在不同组别中的变化非常重要,因为这些变化可以帮助我们去筛选被糖蛋白调控的生物学过程。在PEAKS GlycanFinder中,我们可以实现非标记和标记定量。

非标记定量(LFQ)
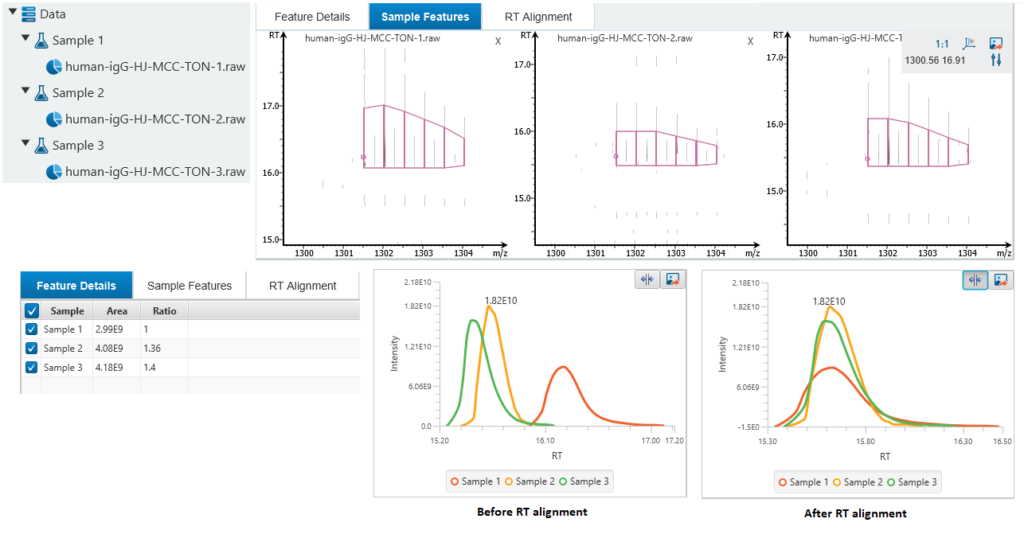
PEAKS GlycanFinder提供非标记定量(LFQ)的工作流,计算蛋白(包括糖蛋白)的相对丰度。定量基于不同样本中肽段特征峰的相对丰度计算,并进行样本间的match between run匹配。
标记定量
不同于LFQ,TMT/iTRAQ的定量是基于MS2或MS3谱中特定m/z处报告离子的相对信号强度实现的。
标记后的混合样品上机时,来源于不同样本的同一条多肽具有相同的母离子m/z和保留时间,同时进行碎裂。在MS2或MS3谱图中,不同样本对应的不同标记试剂产生不同的报告离子,用于计算样本间的相对定量。PEAKS GlycanFinder内置商业化TMT/iTRAQ定量方法,也支持用户自定义标签。

参考文献
[1] Sun, W., Zhang, Q., Zhang, X. et al. Glycopeptide database search and de novo sequencing with PEAKS GlycanFinder enable highly sensitive glycoproteomics. Nat Commun 14, 4046 (2023). https://doi.org/10.1038/s41467-023-39699-5