PEAKS DIA Workflow

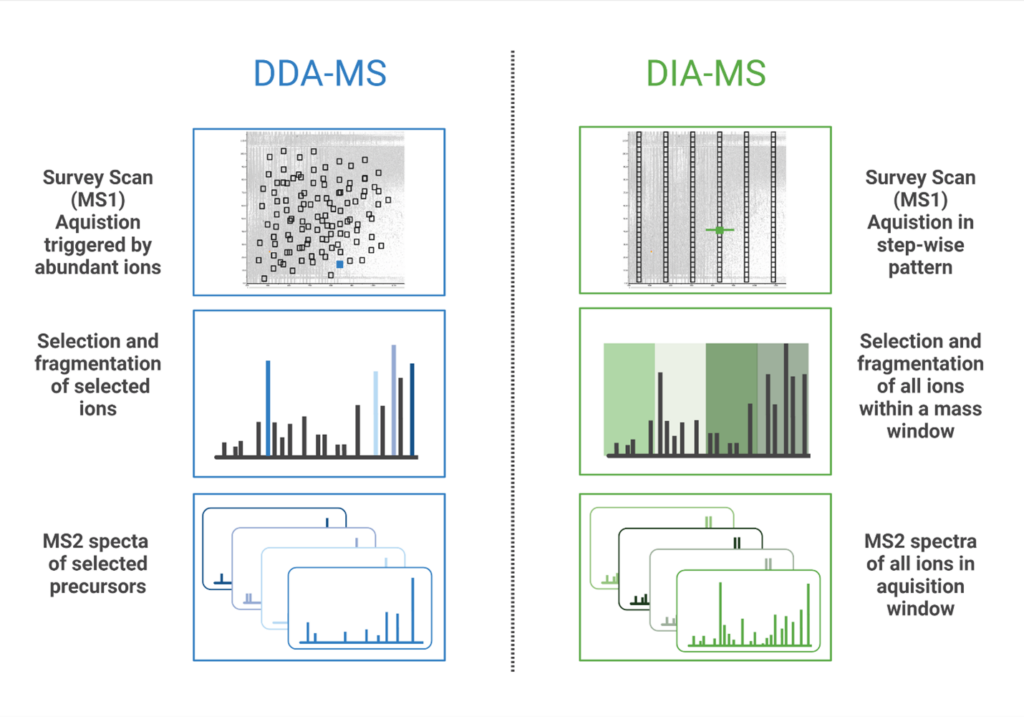
数据独立采集(DIA)是一种在设定的质荷比(m/z)窗口内的所有离子被碎裂和分析的质谱(MS)方法。与数据依赖采集(DDA)相反,DIA工作流可以不局限于最丰富的前体离子而重复识别和量化多肽。在DDA模式下,质谱仪从MS1扫描中选择最高丰度的前体离子,只有这些选择的肽段被串联质谱碎裂并分析。在DIA中,在给定时间内碎裂所有进入质谱仪的离子(broadband DIA)或在固定或可变的m/z窗口内顺序碎片离子 (SWATH(顺序窗口获取所有理论碎裂离子)),从而产生MS/MS 图谱(1)。最近,开发了diaPASEF(并行累加-串联碎片)技术,将捕获离子迁移分离(TIMS)与DIA相结合,以提高肽检测的灵敏度并增加蛋白质组覆盖深度(2)。PEAKS Studio和Online都容纳了上述所有类型的DIA数据,无论采用哪种方法都将有助于分析。
DIA工作流程的优点
• 分析所有肽段,进而减少偏差
• 在MS run中提高的多肽鉴定和定量的重复性(3)
• 在动态范围内定量复杂混合物中的蛋白质
• 消除抽样不足的影响
• 增加蛋白质组覆盖的灵敏度和深度
• 与DDA相比,精度和可重复性更高(4)
• 通过非标记定量消除成本和时间
NEW PEAKS DIA Workflow
PEAKS 12 新功能
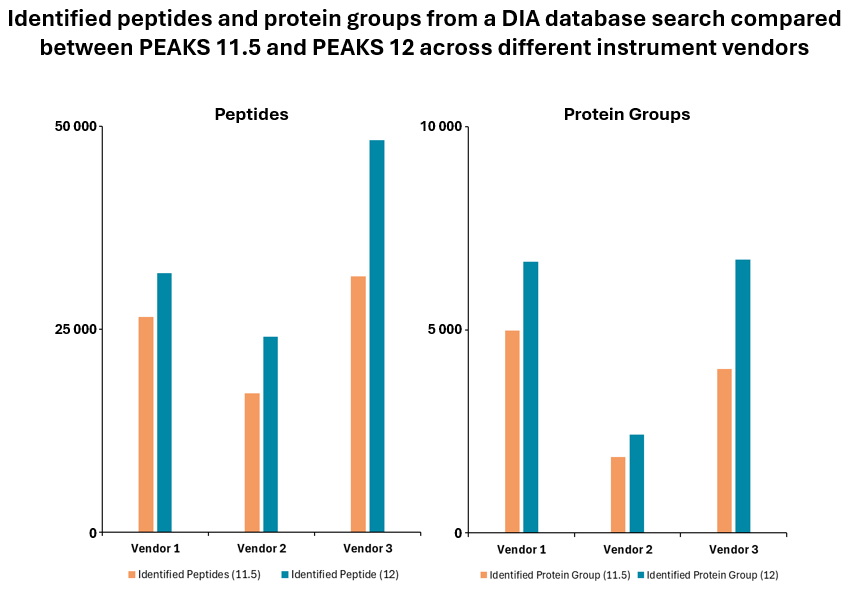
>通过算法的改进,使肽段和蛋白质的鉴定数提高了20%;
>在一个软件包中完成发现和靶向蛋白质组学的完整解决方案;
>用于DIA的DeepNovo肽组学工作流程
>可选Match Between Runs步骤
>支持MSFragger谱库格式
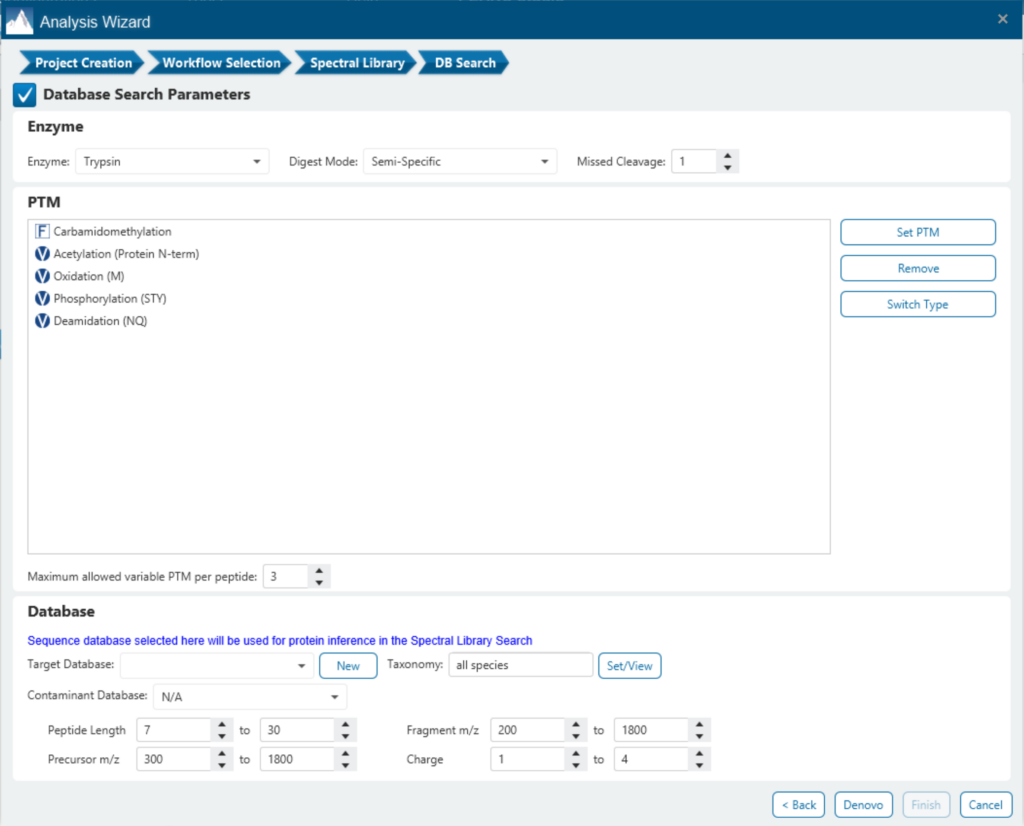
使用PEAKS DIA工作流分析DIA数据集最大化提高鉴定率
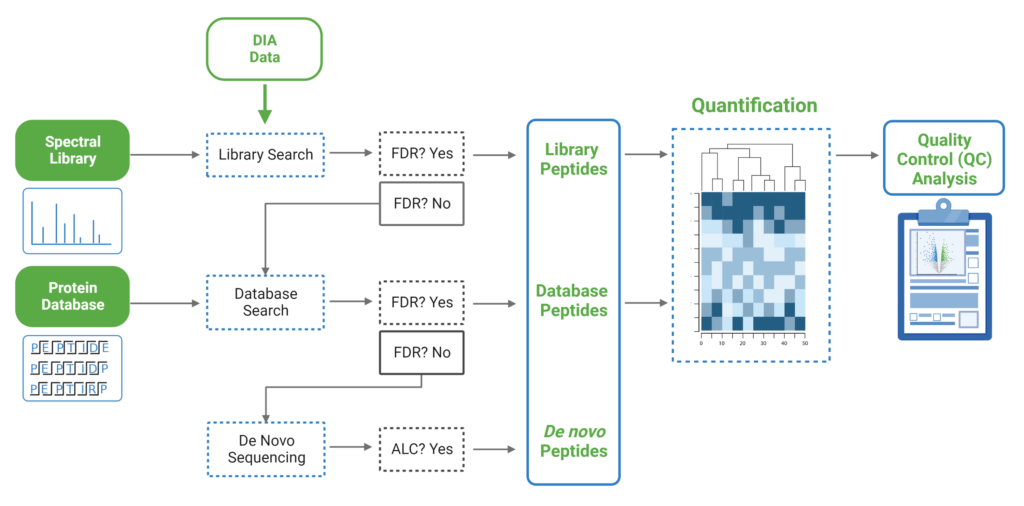
由于DIA MS/MS光谱是由一个m/z窗口内的一系列母离子产生的,它们通常包含来自多个多肽产生碎裂的信号。这对从DIA光谱中多肽鉴定算法提出了独特的挑战。PEAKS 12为解决这个问题提供了一个强大而灵活的解决方案,通过使用谱库搜索(无论是否包含用于蛋白质推断的序列数据库)、直接序列数据库搜索(direct Database)或几种方法的组合来进行肽定性和定量,以获得蛋白质组深度的肽段覆盖(5)。此外,可以应用从头测序来解析那些没有得到很好匹配的谱图。
对于谱图库和直接数据库搜索,搜索空间是放大的。首先,对先前鉴定获得的谱图库进行数据库搜索,通过错误发现率的阈值设定,保留通过筛选的多肽。在FDR阈值内没有得到良好匹配的谱图,进行直接数据库检索。来自与数据库匹配的可靠肽段会添加到结果中。然后,使用相同的FDR阈值设置,使用从头测序分析在数据库搜索中未匹配的谱图。
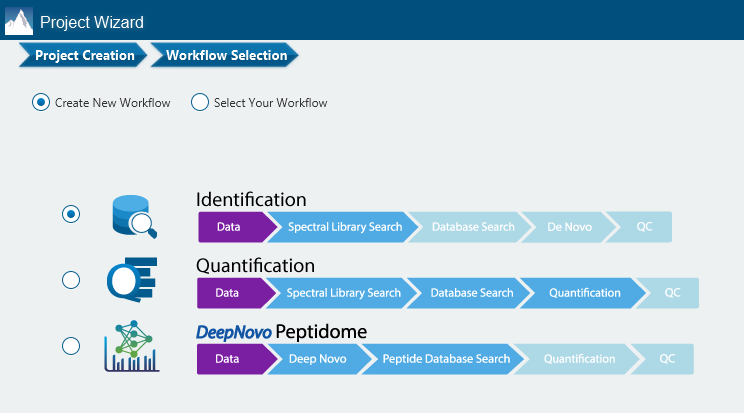
DIA数据有几个可用的工作流程。传统上,常用的工作流程是从DDA数据生成一个库,并在搜索DIA数据时使用这个库,使得数据库搜索可以识别更多的多肽。另一种工作流程是使用PEAKS 12直接搜索DIA数据 (不需要事先构建谱库),进一步对于未匹配的图谱可以进行从头测序,另外在定量模块中,可以对DIA数据进行非标记的定量。在 PEAKS12中,QC分析提供了定性定量的数据质控的统计概要和可视化结果。 Learn more
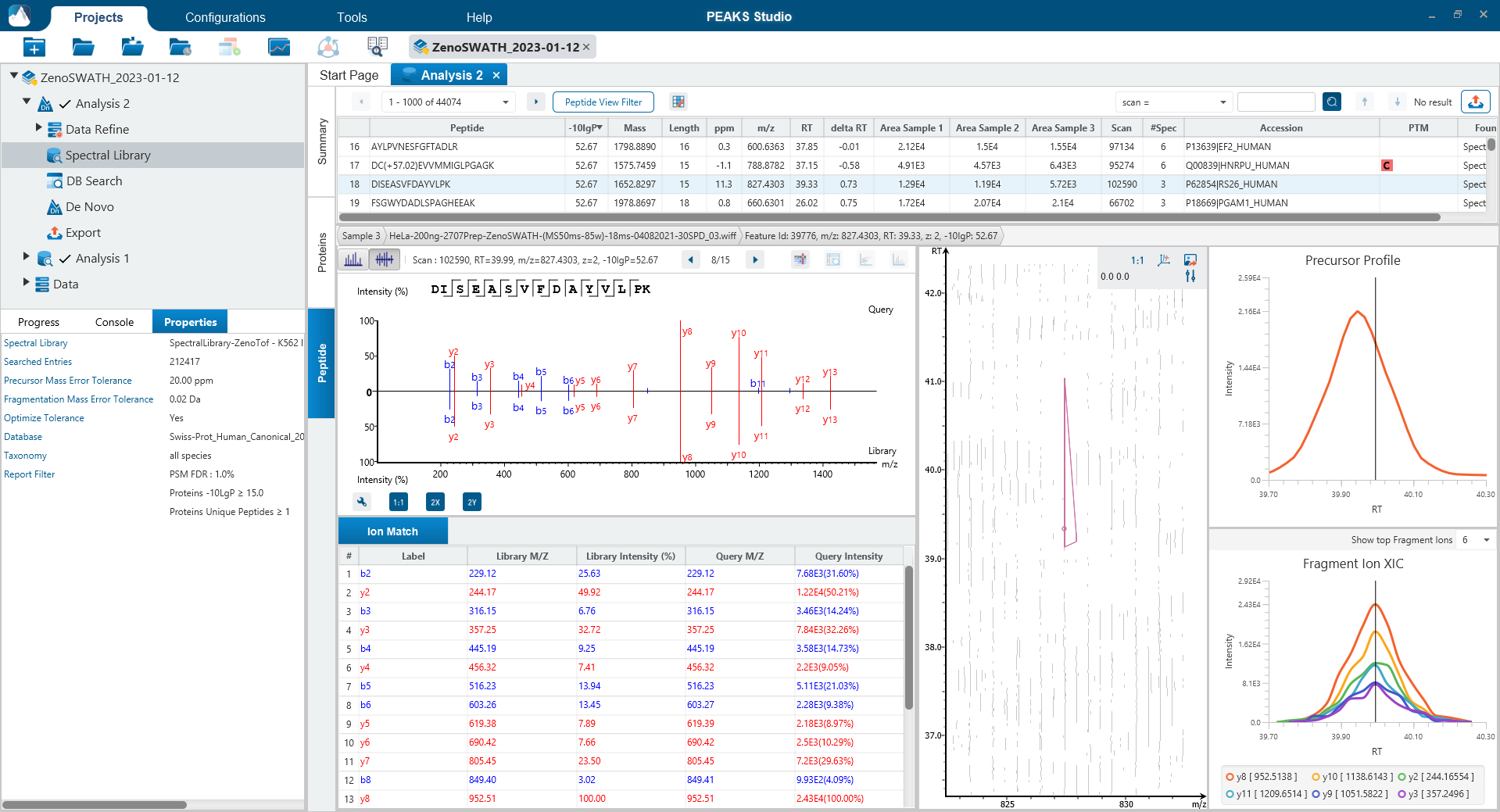
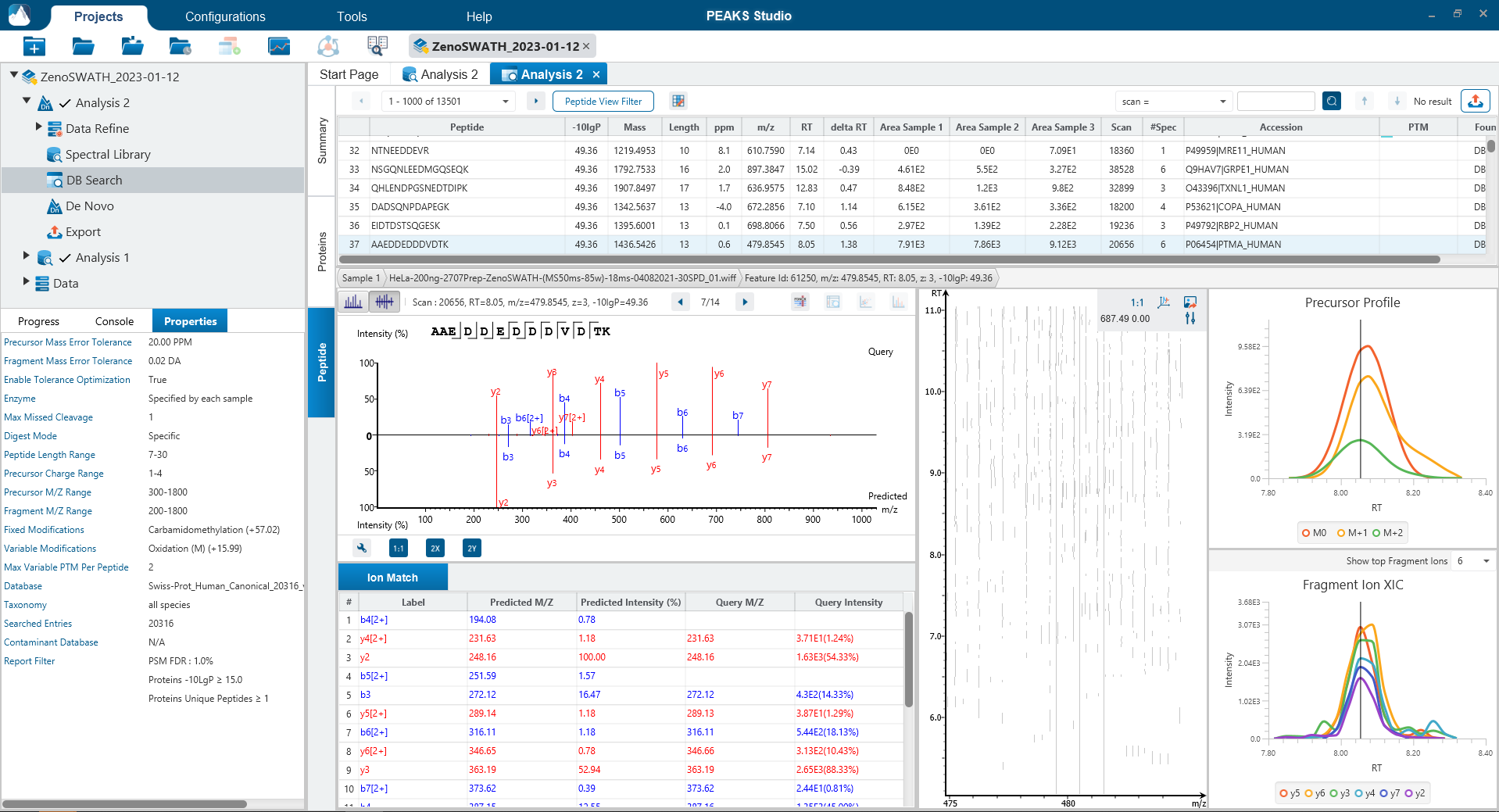
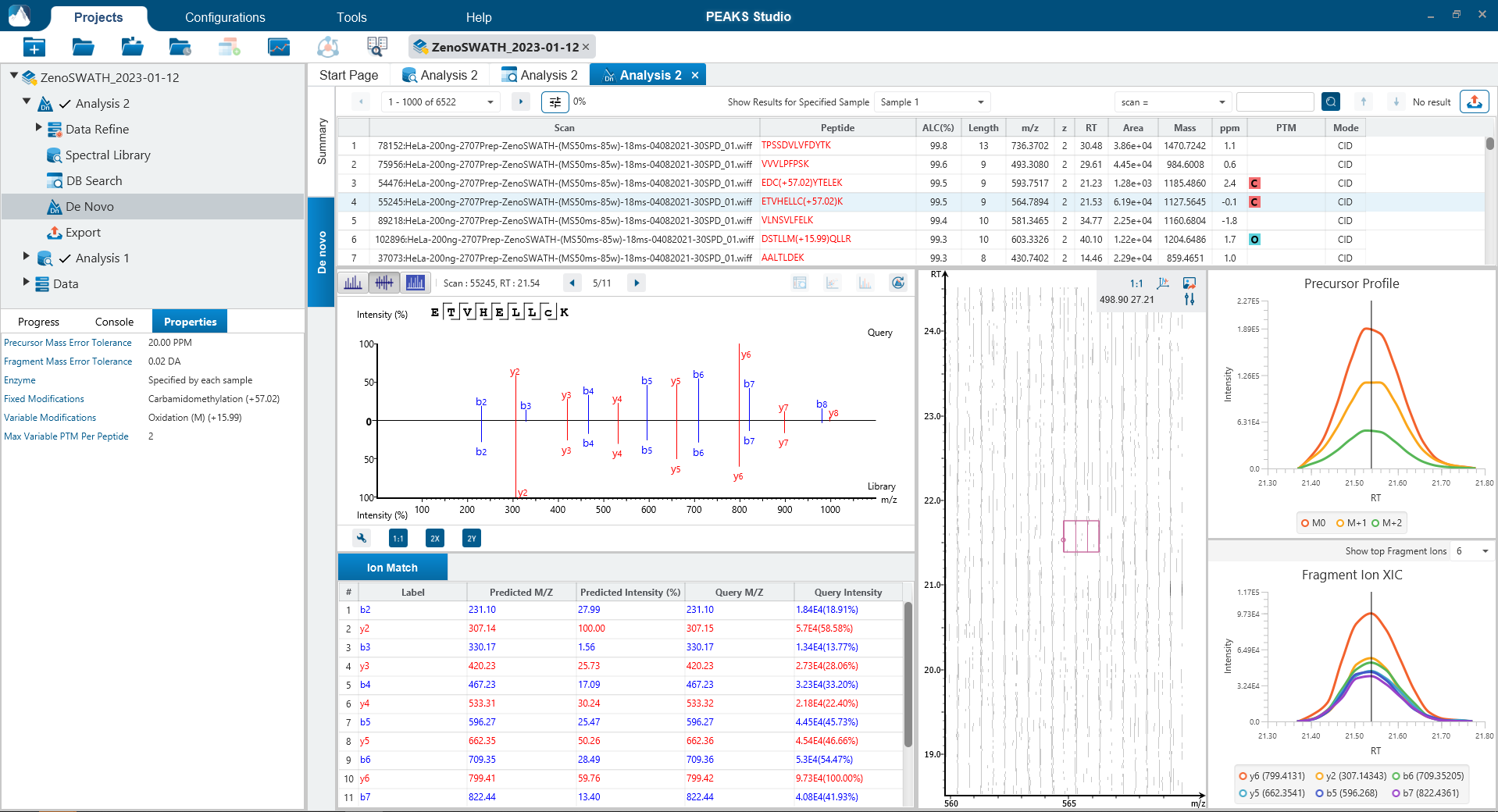
可选Match Between Runs步骤
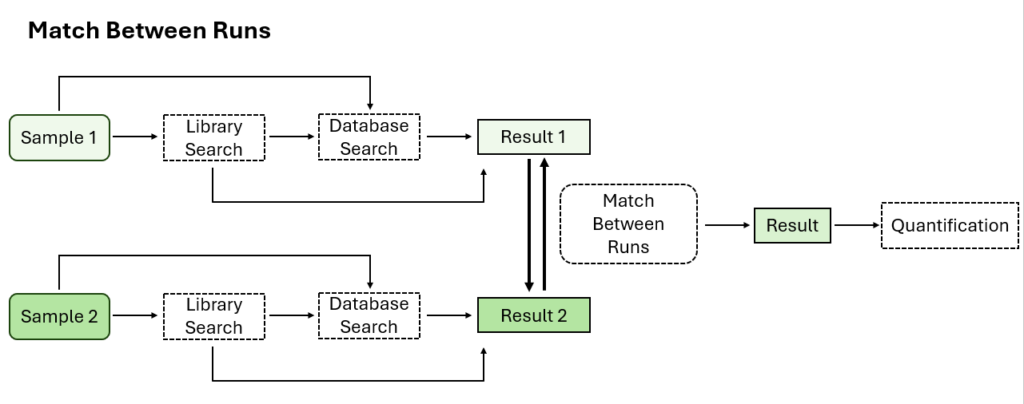
Match Between Runs允许基于电荷状态、m/z、保留时间和离子迁移率对多个样本的特征进行对齐。该方法允许在一个样品缺少MS2鉴定的多肽的情况下将另一个样品中MS2鉴定到的多肽转移到该样品,以增加覆盖率。在以前的PEAKS版本中,match between runs是在定量步骤中完成的 。现在在PEAKS12中,用户可以选择在DIA工作流的鉴定步骤执行match between runs。
在DIA工作流中全面支持PTM分析
由于巨大的搜索空间和DIA谱图本身的复杂性,在直接序列数据库搜索中,对于翻译后修饰的鉴定有很大的挑战。通过重新训练深度学习模型,并且在计算中引入翻译后修饰的化学分子式作为通用代码,PEAKS 12 DIA工作流现在支持鉴定用户指定的任何PTMs(包括用户自定义翻译后修饰)。这将增加对修饰肽的鉴定,而不需要在谱图库中存在相应的修饰肽段的条目。
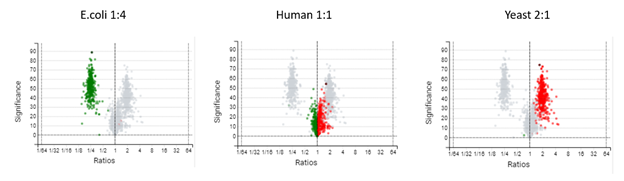
定量具有更高的重现性
与DDA方法相比,DIA质谱产生高度可重复性的定量结果。谱库和直接数据库搜索结果可以结合非标记量化方法。为了提高准确性,与母离子的XIC profile具有高度相关性的碎片离子纳入在多肽丰度计算中。因而可获得一组具有最小缺失值的稳健的定量结果,以帮助发现不同条件下的生物学显著变化。
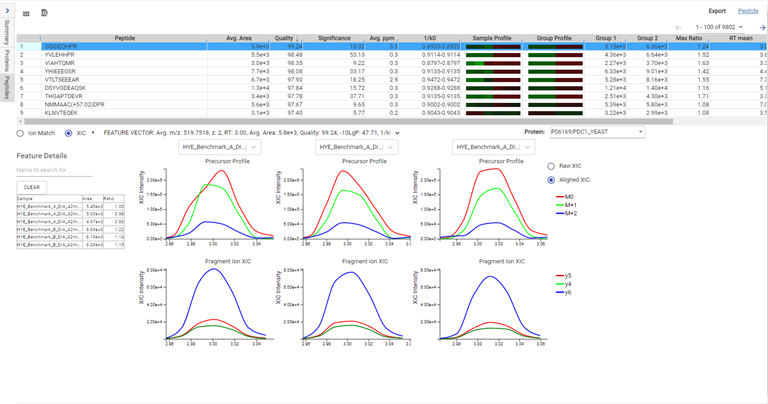
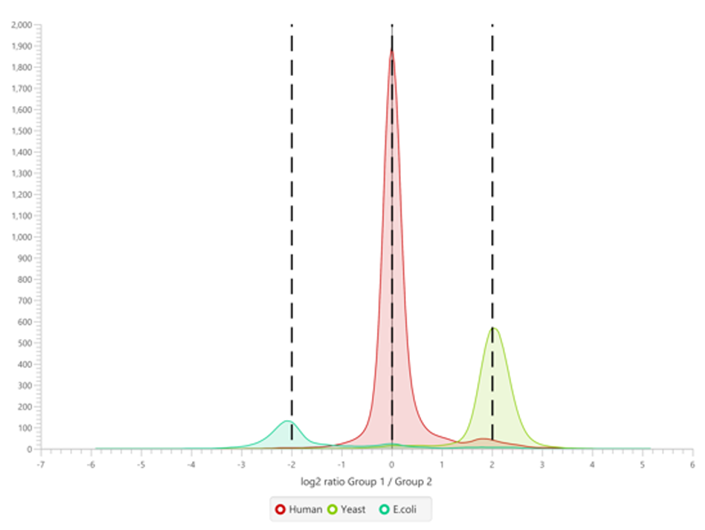
Label-Free Quantification
在DIA LFQ工作流程中,用户在定量模式上有两种选择,优先考虑高精度(更高灵敏度)或高准确度。

参考文献:
- Ludwig, C., Gillet, L., Rosenberger, G., Amon, S., Collins, B. C., and Aebersold, R. (2018). Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Mol. Syst. Biol. 14:e8126. doi: 10.15252/msb.20178126
- F., Brunner,A.D., Frank,M., Ha,A., Bludau,I., Voytik,E., Kaspar-Schoenefeld,S., Lubeck,M., Raether,O., Bache,N., et al. (2020) diaPASEF: parallel accumulation–serial fragmentation combined with data-independent acquisition. Nat. Methods, 10.1038/s41592-020-00998-0.
- Collins, B. C., Hunter, C. L., Liu, Y., Schilling, B., Rosenberger, G., Bader, S. L., et al. (2017). Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Nat. Commun. 8:291. doi: 10.1038/s41467-017-00249-5
- Fernández-Costa, Carolina, et al. “Impact of the Identification Strategy on the Reproducibility of the DDA and DIA Results.” Journal of Proteome Research, no. 8, American Chemical Society (ACS), June 2020, pp. 3153–61. Crossref, doi:10.1021/acs.jproteome.0c00153.
- 05. Xin, L., Qiao, R., Chen, X. et al. A streamlined platform for analyzing tera-scale DDA and DIA mass spectrometry data enables highly sensitive immunopeptidomics. Nat Commun 13, 3108 (2022). https://doi.org/10.1038/s41467-022-30867-7