NEXT GENERATION

de novo Sequencing | Database Search | Spectral Library Search
Accurate and Sensitive Proteomics LC-MS/MS Data Analysis Software for both DDA and DIA
New Features
- Deep learning-boosted ID for PEAKS DB
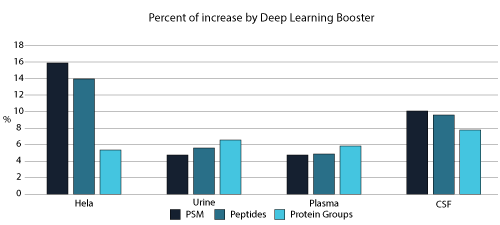
- deep learning-boost can increase the identification over 10% for DDA data analysis.
- Streamlined DIA workflow (Specral Library Search + directDB + de novo)
- Improved identification rate over 30% with Spectral Library and direct database search
- Enhanced Deep learning-based technology for predicting spectra, retention time, and collision cross-section values for Direct database search to improve sensitivity and accuracy including DIA with short gradient
- Improve the completeness of proteome by de novo sequencing
- DeepNovo-based peptidome workflow for immunopeptidomics.
- Integrate homology search with de novo to improve HLA peptidomics with FDR control
- Glycan module – a structure-based solution to glycoproteomics (Available in PEAKS Studio 11)
- Provides structural information of the glycans attached to peptides.
- Locating the glycosylation site.
- Glycan chart visualises the glycans area percentages in a pie chart.
- Quality Control (QC) Function for large cohort DDA and DIA (Available in PEAKS Online 11)
- Confirm experimental consistency
- Ensure data quality
- Validate identification and quantification consistency


PEAKS® is a specialised tool that offers unrivalled peptide identification rates driven by deep learning technology and integrated with library search, direct database search and de novo sequencing. PEAKS® 11 supports data-dependent and data-independent acquisition analyses (DDA and DIA respectively) and ion mobility mass spectrometry (IMS-MS). As a vendor-neutral computing platform, PEAKS® is capable of directly loading raw mass spectrometry data and standard data formats. Deploy the PEAKS® workflows to identify the presence of peptides and proteins in your project for 1) DDA data analysis including de novo sequencing, PEAKS DB (database search) identification, PEAKS PTM (post translational modification) analysis, SPIDER homology search and PEAKS DeepNovo Peptidome (specialised workflow for peptidomics data) and 2) DIA data analysis including spectral library search, direct database searching and de novo sequencing.
Quantification analysis by labelling and label-free quantification (LFQ) can also be performed using the PEAKS Q add-on module. Intuitive result visualisation tools are provided at every stage of analysis and results can be exported.
PEAKS® DIA Workflow
PEAKS® offers a robust solution for DIA data analysis. It incorporates three methods of peptide identification: spectral library search, direct database search, and de novo sequencing. The search is performed using an expanding search space. First, a library search is performed against a library of previously identified spectra. By predicting the false discovery rate, peptides that pass the filter are saved. MS/MS spectra that don’t match a peptide within the false discovery rate threshold are brought forward to a direct database search. Confident database matches are added to the result. Then, using the same FDR approach, unmatched spectra from the database search are analysed using de novo sequencing.
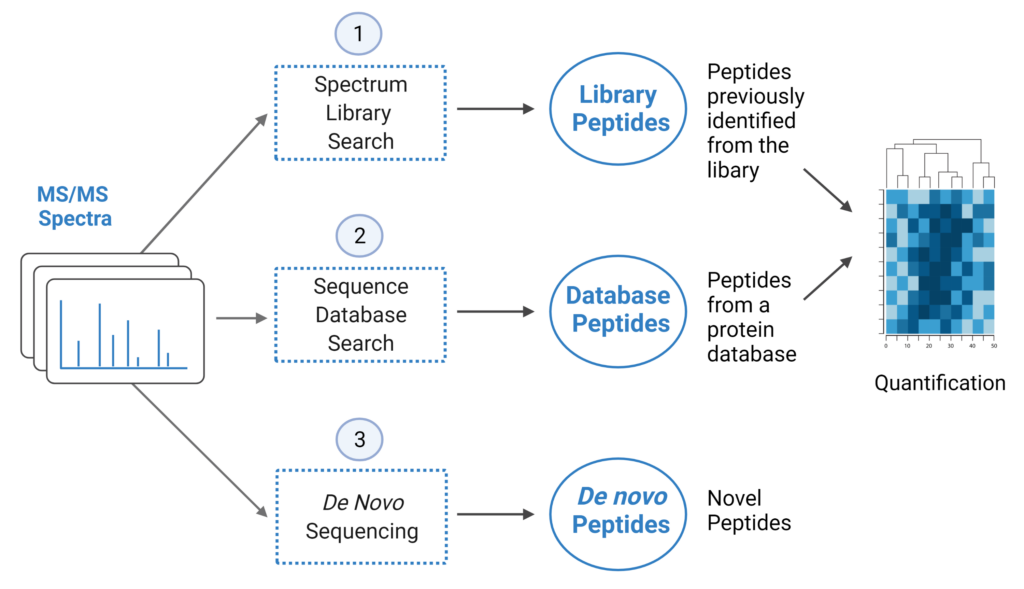
- A library search is performed against a pre-defined spectral library. Peptide spectra without a library match can be directly searched against a protein database.
- A protein sequence database is directly searched with DIA data. Advanced machine learning algorithms allow improved accuracy and sensitivity of peptide identification.
- Unmatched spectra from the database search are de novo sequenced.
- Identified peptides from both the spectrum library search and protein sequence database search can be used in a quantification analysis.
Note: All 3 search methods, PEAKS Spectral Library Search, Direct Database Search, and de novo sequencing are optional, and the steps of the workflow can be conducted independently or in sequence.
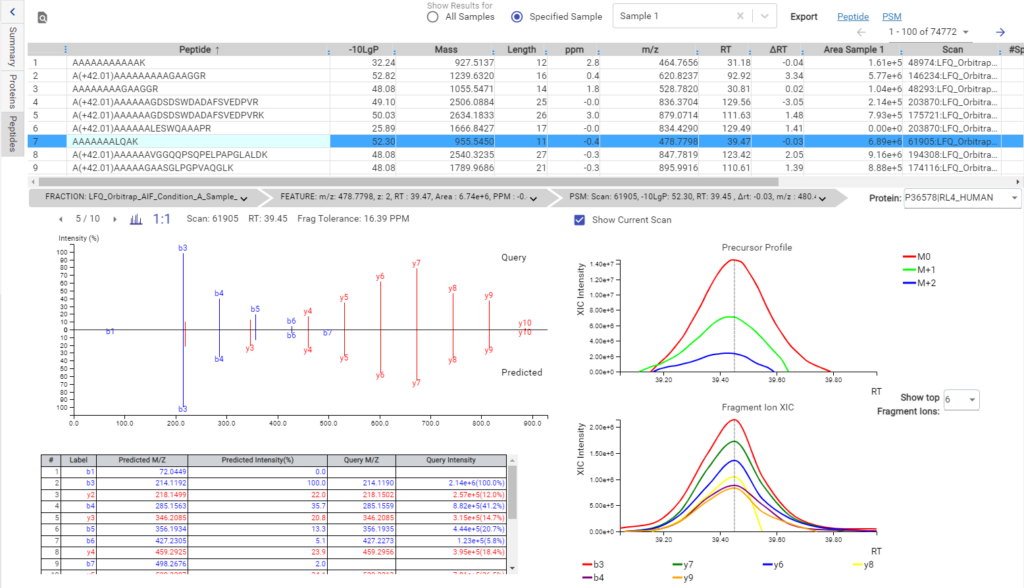
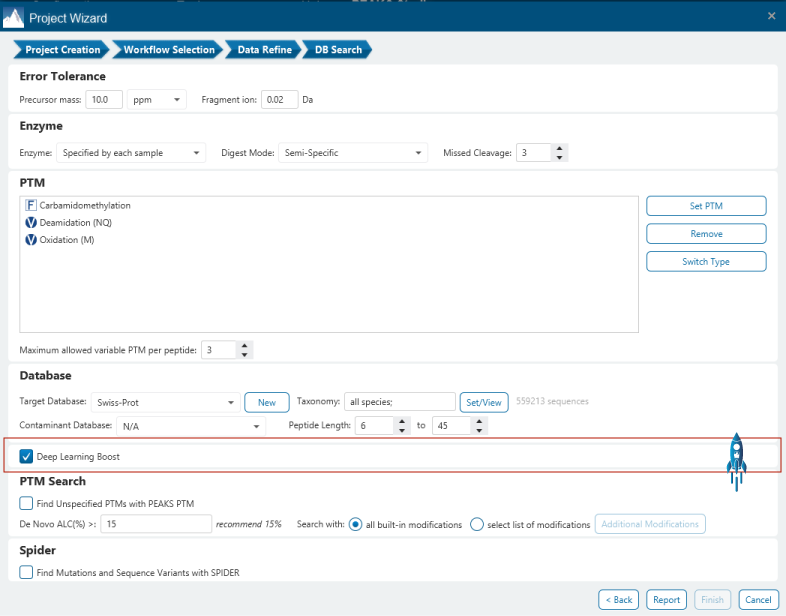
Deep learning is utilised in DDA workflows in the PEAKS de novo-assisted database search to boost PEAKS DB search results.
From testing different sample types, tissues, and multiple MS instruments, deep learning-boost can increase the identification over 10% for DDA data analysis. Artificial intelligence provides enormous opportunities to accelerate proteomics research through comprehensive peptide and protein identification and quantification.
This newly developed solution is a specialised workflow for peptidomics data that combines database searching, de novo sequencing, and identification of mutated peptides.
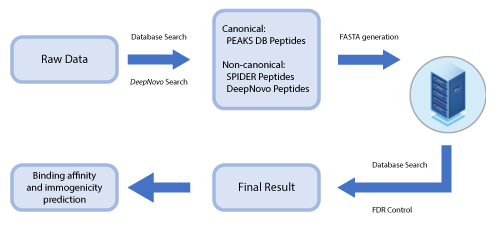
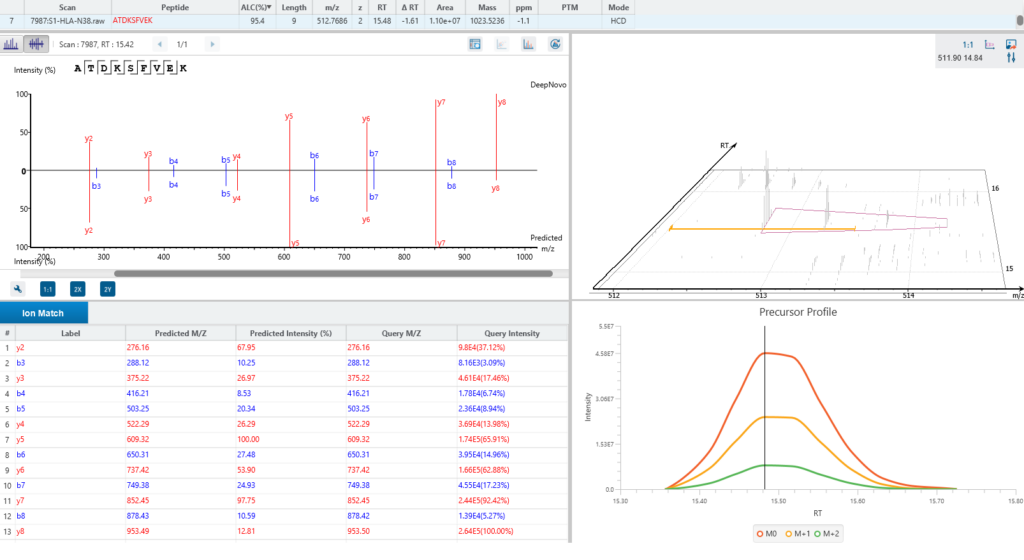
By training DeepNovo deep learning model using peptidomics datasets the sensitivity and accuracy of peptide identification can significantly be improved. Furthermore, de novo peptides (non canonical) are combined with database peptides (canonical) for more accurate estimation of false discovery rate.
As an optional add-on module, PEAKS Glycan Module add-on enables scientists to determine glycan site localisation and glycan structures.
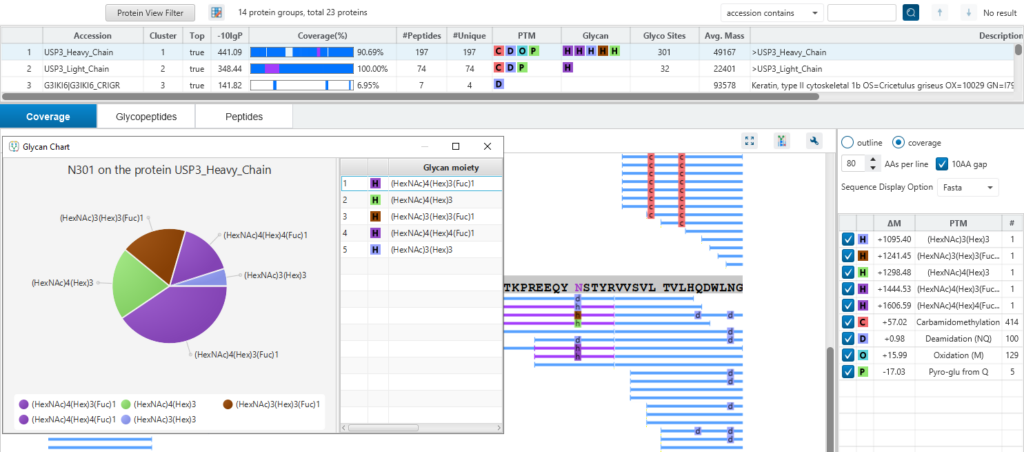
- Provides structural information of the glycans attached to peptides.
- Locating the glycosylation site.
- Glycan chart visualises the glycans area percentages in a pie chart.
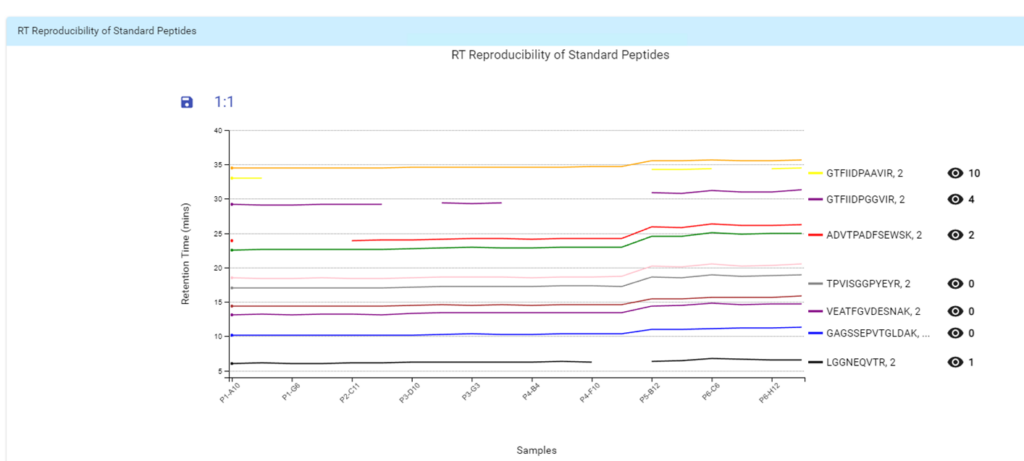
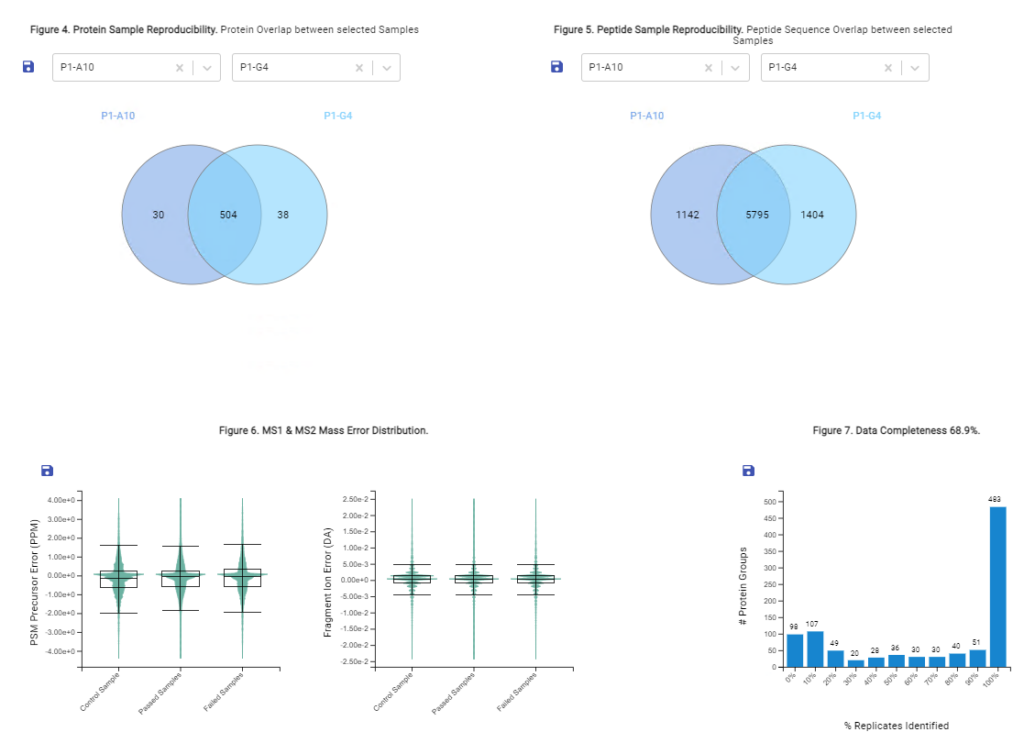
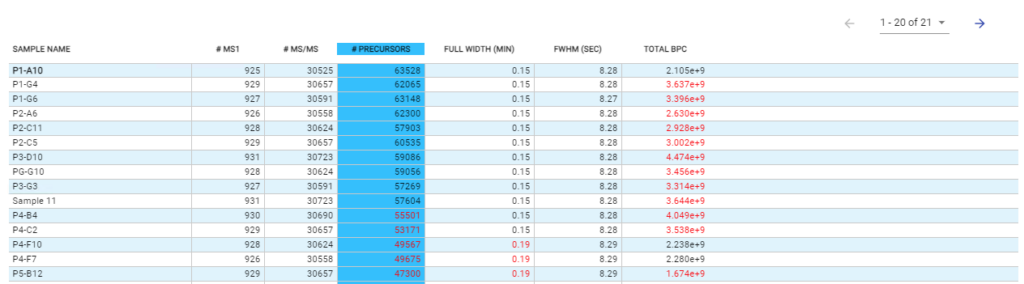
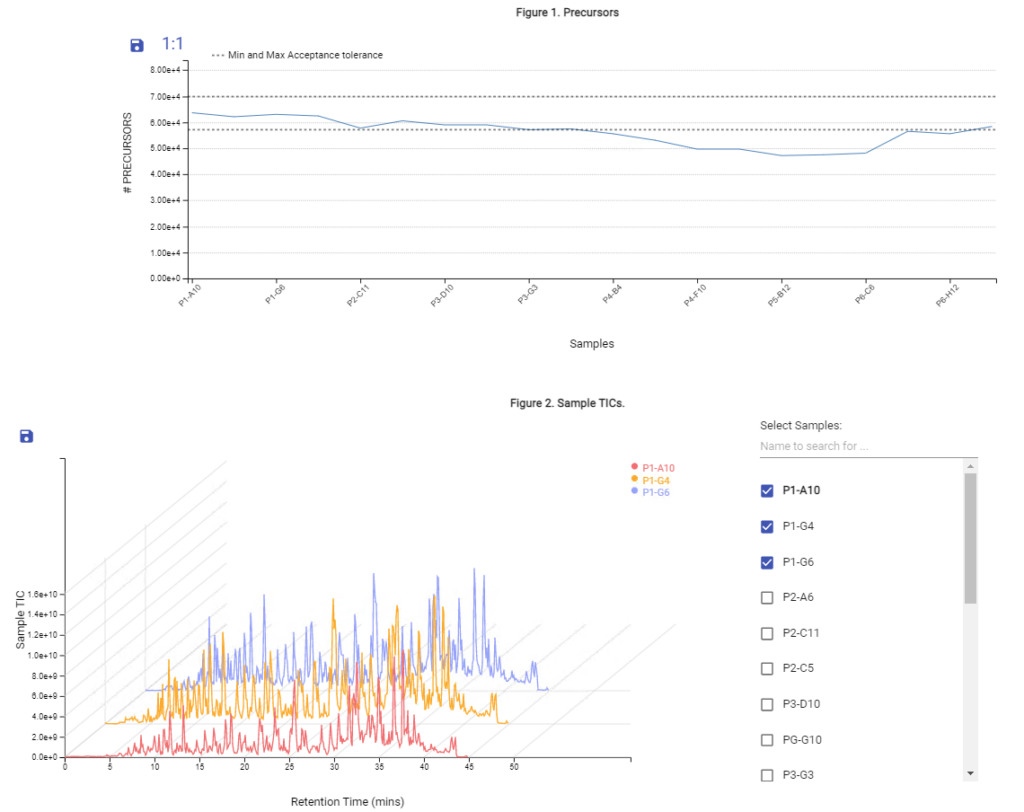
With the new Quality Control (QC) analysis in PEAKS® Online 11 users can assess statistical information of the raw data and/or results and gain beneficial insight into the attributes of the LC-MS acquisition. This automated tool is designed for both, DDA and DIA data and will supply the elements to determine the quality of the data and evaluates the setup of the experiment.