 de novo Sequencing Intro
de novo Sequencing Intro
背景介绍
多肽从头测序(de novo sequencing)是指在不借助序列数据库的情况下,通过串联质谱(MS/MS)得到多肽氨基酸序列的分析过程。这与另一种流行的肽段鉴定方法–“数据库搜索”形成了鲜明对比,即在给定的数据库中搜索以找到目标肽。从头测序的一个明显优势是它既适用于数据库搜索,也适用于新肽。
基本原理
在串联质谱中,质谱通过对肽链碎裂产生的碎片离子检测,进而产生ms/ms谱。根据所采用的碎裂方法,可以产生不同的碎片离子类型。目前应用最广泛的碎裂方法是碰撞诱导解离(CID)和电子转移解离(ETD),CID主要产生b和y离子,ETD主要产生c和z离子。
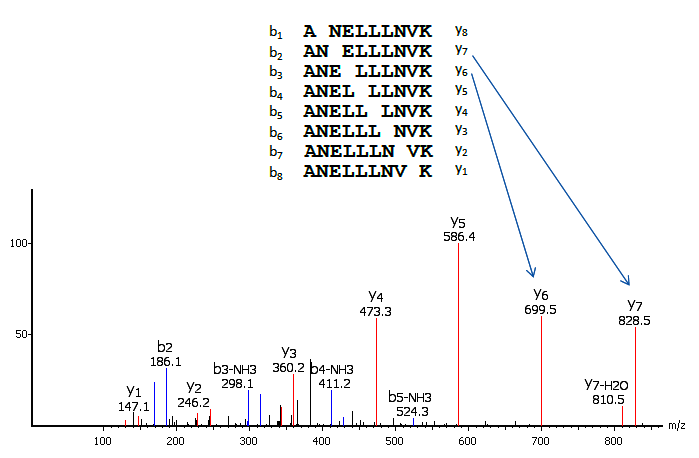
de novo sequencing的主要思路是用两个碎片离子之间的质量差值来计算肽主链上氨基酸残基的质量。质量数通常可以独特地确定残基的类型。例如,在图1中y7和y6的质量差值为129,即E的残基质量数。同理,下一个相邻的残基,可以通过y6和y5之间的质量差确定为L。该解谱过程会进行下去,直到所有的氨基酸残基都被确定。
因此,如果能在谱图中识别出y离子或b离子序列,就可以确定肽的序列。然而,质谱仪获得的谱图并不能告诉我们峰的离子类型,这需要专家或计算机算法在从头测序过程中计算出来。一些因素会给这一过程带来困难,比如:
- 不正确的b、y离子分配
- 一些离子碎片的缺失(比如图1中的 b1 、 y8 )
- 谱图中其他碎片离子类型(比如图1中的 b3-NH3)
- 谱图中存在噪音峰
- 高度相似的氨基酸残基质量数,(I=L and K=Q)。
- 残基上的PTM(翻译后修饰)可能导致质量模糊,并使肽碎片类型复杂化。
以上这些因素,可能导致de novo测序只能从谱图中计算出部分正确的序列标签。
自动化的de novo测序
手动进行de novo测序的解谱工作要求人员的专业性,并且十分地费时。一个可靠的自动化de novo测序解决方案可以节省宝贵的人工时间,并大大降低了实验室的人工成本。自动化de novo测序在生物信息学领域得到了广泛的研究,并开发了多种算法,虽然计算机算法所使用的基本原理与人工de novo测序相同,但计算机算法的执行过程通常与人工分析不尽相同。
PEAKS Studio软件于2002年首次发布,目前已成为自动化从头测序的行业标准软件,并以其准确性、快速分析的能力,以及用户友好性而闻名。
结合数据库搜索使用
过去,人们常认为de novo测序分析的速度非常慢。因此,只有在蛋白质数据库不可用的情况下,才使用de novo测序的方法。然而,随着计算机算法(如PEAKS)的最新发展,速度已不再是问题。这使得在蛋白质组学研究中对得到的每一张质谱图都进行de novo测序成为一种可行的选择。即使是当数据库可用的情况下,de novo测序也可以通过以下方式帮助多肽的鉴定。
- denovo测序肽段与数据库搜索肽段之间的匹配或相似度可以很好地说明数据库搜索结果的正确性。因此,从头测序可用于提高数据库搜索性能。
- de novo测序得到的肽段如果在数据库中没有显著匹配,可能是在样品中存在全新的肽段,值得进一步确定,有可能发现预期外的PTM或者多肽的突变。
如下图2所示,PEAKS Studio软件在设置标准操作流程的时候,已经考虑到了这个问题。

- Tran NH, Qiao R, Xin L, Chen X, Liu C, Zhang X, Shan B, Ghodsi A, Li M. Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nature Methods. 16(1), 63-66. 20/12/2018.
- Hughes, C., Ma, B., Lajoie, G.A. De novo Sequencing Methods in Proteomics. Methods Mol Biol. 2010;604:105-21.
- Ma, B. & Johnson, R. De novo Sequencing and Homology Searching. Molecular & Cellular Proteomics. 2012;11(2).