
PEAKS AB 3.0新版本横空出世!算法全面升级从头测序功能,从抗体测序飞跃到任意蛋白测序,整合Intact Mass数据验证测序结果。在从头测序数据中提供糖基化位点的全景表征。
PEAKS AB使用液相质谱(LC-MS/MS)多酶联合酶解的数据集中提供蛋白水平的从头测序自动化方案。从高置信的从头序列标签开始,使用de Brujin加权算法组装完整的蛋白质序列。
# 新功能 #
在PEAKS AB 3.0中,提供了全新的功能提高测序准确性和结果呈现,包括:
- 非抗体蛋白的序列组装
- 深度的聚糖全景分析
- 手工编辑和查找同源序列
- 先进的完整分子量解卷积分析
- 支持ZenoTOF仪器的数据
深度的聚糖全景分析
在PEAKS AB 3.0中引入的一个新功能是Glycan Profiling工具。该工具对抗体重链和/或轻链中鉴定的N-糖基化位点进行深入的聚糖分析。除了精确的糖肽映射到组装的抗体序列之外,基于酶的聚糖谱学分析显示了在选定的糖基化位点上每个聚糖的组成和相对丰度。在每个糖肽谱中提供了聚糖组成和结构注释,并基于与N-链接糖基化数据库匹配聚糖片段离子。
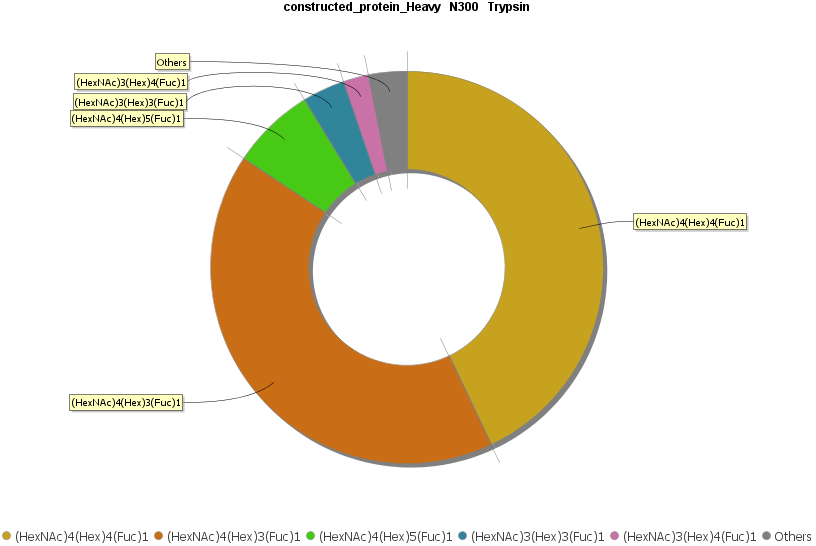
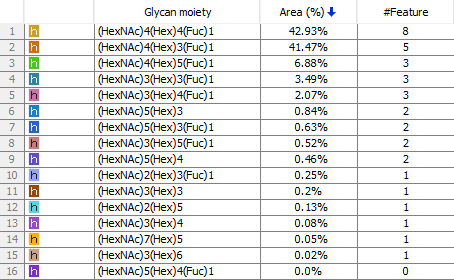
非抗体蛋白从头测序组装
在PEAKS AB 3.0中,除抗体外的其他蛋白样品也可以测序。勾选“使用参考序列(提供一个或两个FASTA序列)”后的方框,将目标序列添加到下面的白色方框中。
在蛋白测序中,de novo标签用于组装未知序列,而提供的参考序列,可用于填补测序的缺失部分。
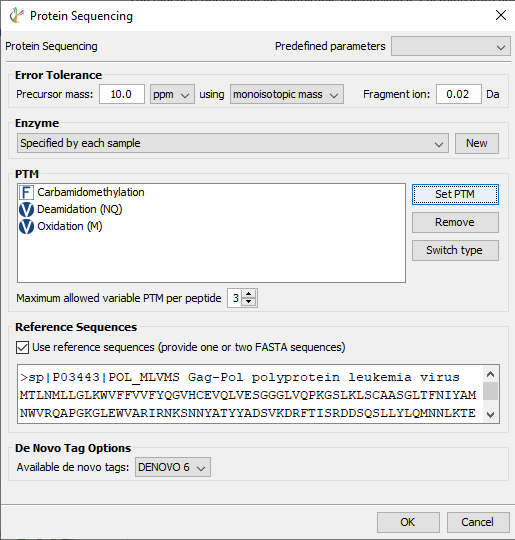

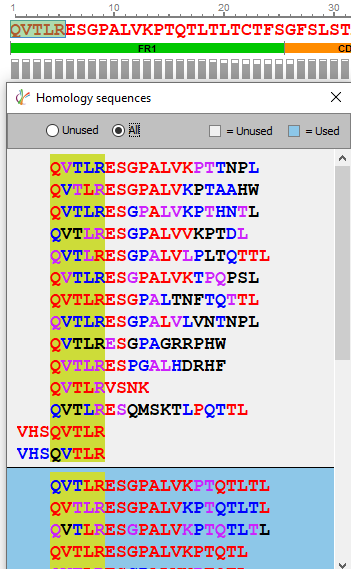
手工编辑和查找同源序列
序列验证和手工编辑是任何蛋白质测序项目的重要步骤。测序错误可能是由于同源蛋白的背景污染或低覆盖率和数据质量不好引起的。通过选择3个或更多的氨基酸,用户现在可以选择“查找同源序列”来显示任何可以映射到该区域的多肽序列可以用于替换。
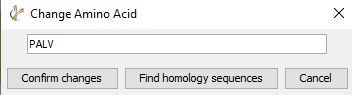
显示的备选多肽序列根据每个氨基酸的局部置信度评分进行颜色编码,以便快速评估多肽序列的结果质量。通过选择待编辑的氨基酸,然后更改为修改后的序列,点击“确认更改”将编辑参考序列。在第二轮序列组装后,从peptide mapping视图中点击“apply”,应用这些更改将生成一个新的测序结果节点。
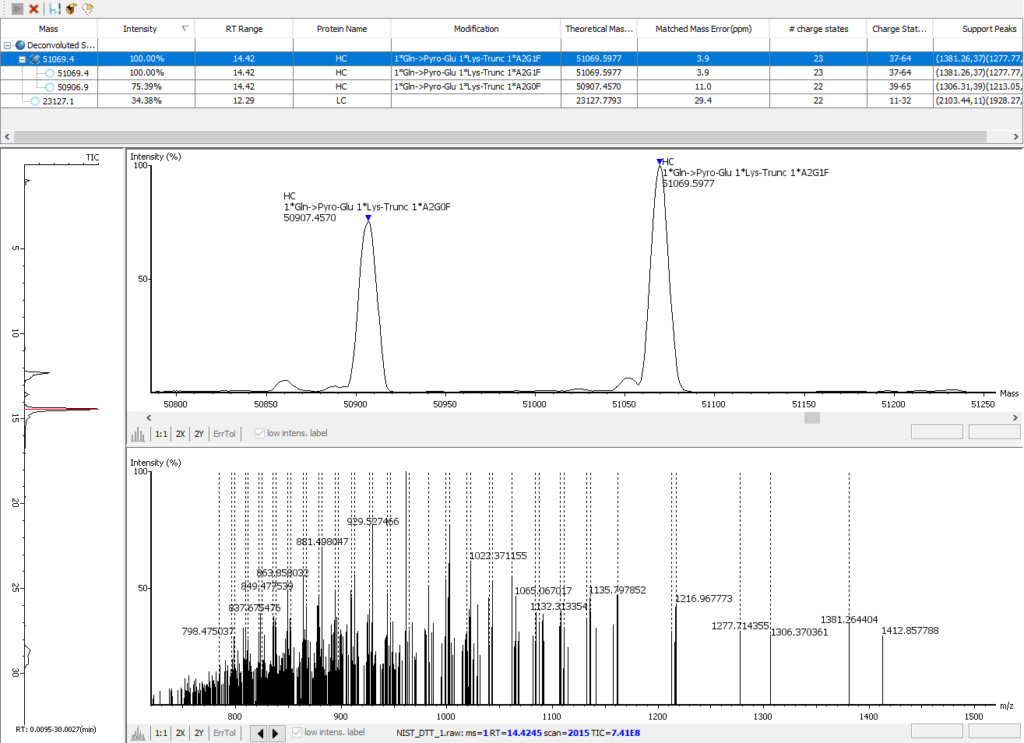
优化的完整分子量解卷积分析
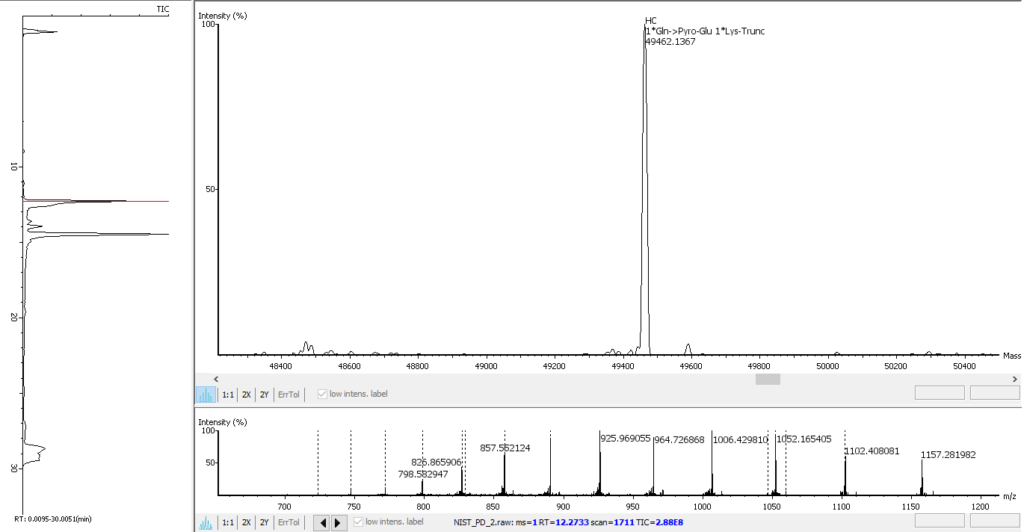
PEAKS AB 3.0先进的解卷积算法可实现自动化、高效和准确的完整分子量分析。对蛋白质进行完整分子量的检测,以验证序列组装的正确性以及判断任意修饰的个数,如N端焦谷谷氨酸(Gln->Pyro-Glu),C端赖氨酸截断(1 * Lys-Trunc)和N-链接糖基化(如1 * A2G0F),如下图所示。
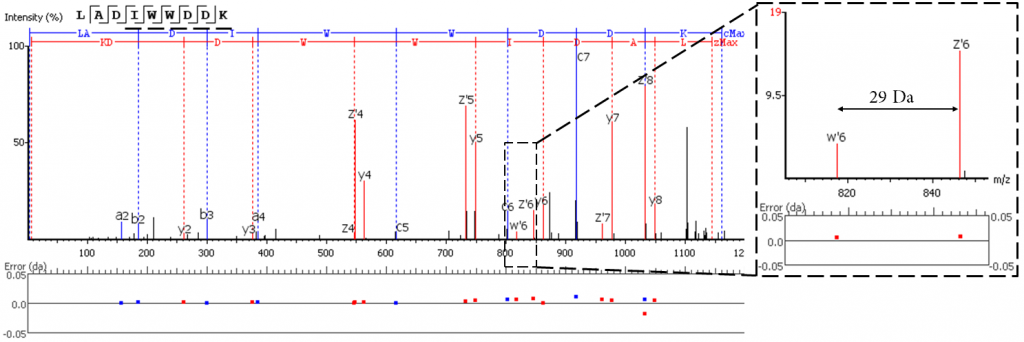
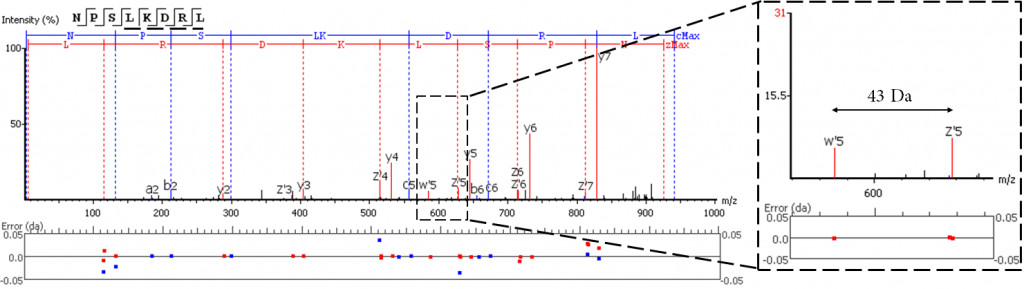
强化亮氨酸和异亮氨酸三重水平的区分
EThCD或EAD碎片从异亮氨酸的-C2H5 (-29 Da)和亮氨酸的-C3H7 (-43 Da)的特征性丢失中产生标志性的w-ion。PEAKS AB利用这些特征碎片的信息,再加上酶切特异性和同源抗体序列分析,优化了Ile/Leu的区分。在3.0版本中,PEAKS AB加入了对EAD数据的支持,扩充了w-ion的产生来源。
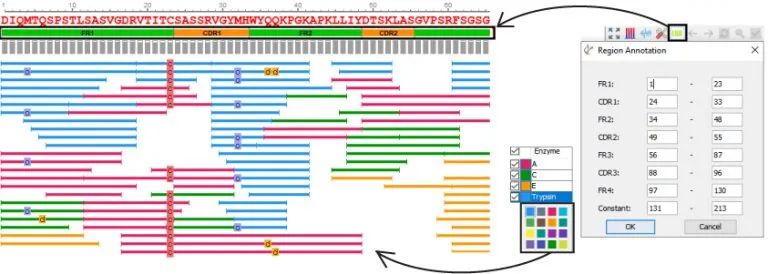
更丰富的用户定制化选择
PEAKS AB的设计是用户友好的,最终可以减少蛋白质从头测序所需的人工工作量。可视化地访问结果为用户提供了PEAKS额外的优势,可以精确地检查结果。
- 定制酶的颜色
- 自动化和自定义CDR注释
参考文献
Tran, N.H. et al. Complete De Novo Assembly of Monoclonal Antibody Sequences. Scientific Reports. 6:31730. 26/08/2016.
Leave a Reply