

基于质谱的多肽组学已成为开发多肽疫苗和免疫疗法的热门方法。 这一研究领域也导致了许多non-canonical多肽的发现,并改变了我们对基因组区域阅读和翻译的理解。在多肽组学中,将正确的氨基酸序列分配到MS/MS谱图是至关重要的,因为每个残基的位置和鉴定结果直接影响多肽-蛋白质的相互作用,并确定它是否是进一步完成下游分析的优质候选。随着多肽组学分析和免疫肽组分析的普及,迫切需要有一个软件工具以准确和直观的方式有效地处理这类数据。
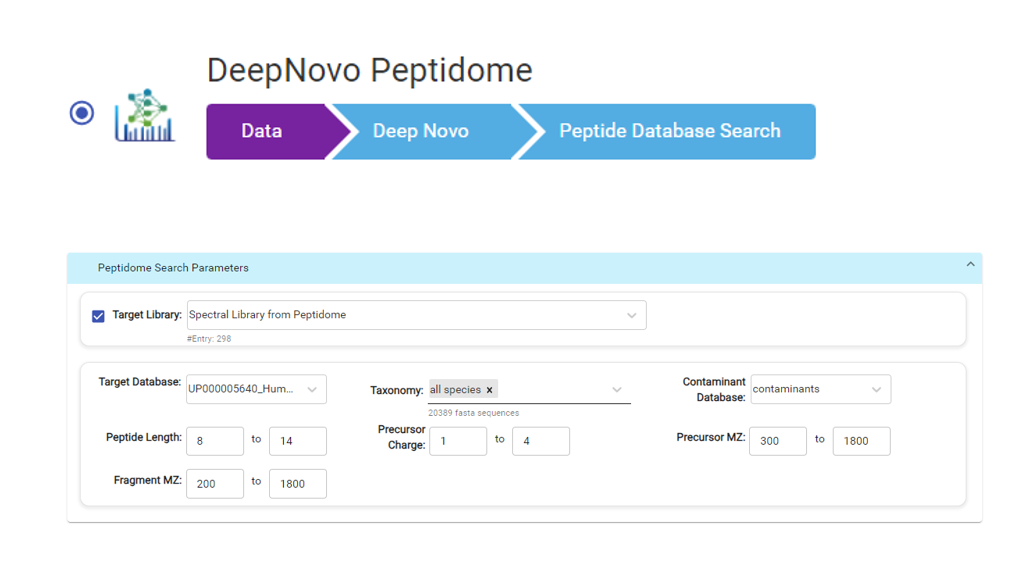
在多肽组学数据的质谱分析中,PEAKS软件已成为首选的数据分析解决方案,作为PEAKS Studio 11和PEAKS Online 11的一部分,我们提供了一个新的DeepNovo Peptidome的肽组学专属工作流程。
功能核心
- 在工作流中整合了 de novo sequencing, 序列数据库搜索, 以及同源搜索
- 为多肽的de novo测序结果提供全局的FDR评估
- 深度学习模型经过了大规模HLA多肽数据集对保留时间、碎片离子和离子淌度预测等参数的训练
- 考虑三大类翻译后修饰,更准确地预测修饰肽段的保留时间和MS2谱图。
- 通过增加Non-Canonical数据库搜索改善了DDA peptidome
- 通过Gene tab 整合了蛋白组与基因组信息
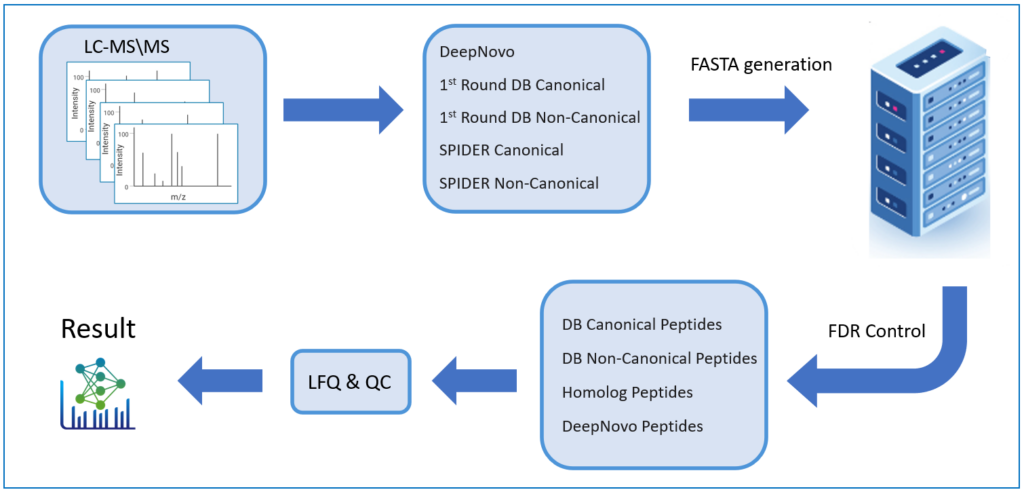
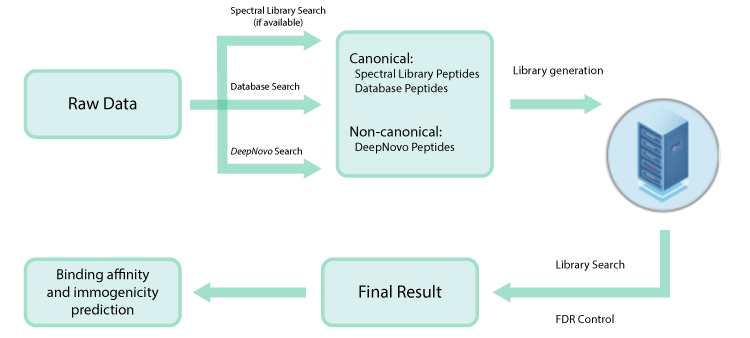
以前,对于多肽的从头测序结果,无法进行FDR评估,因此通常用户会选择一个ALC的打分阈值来过滤低质量的结果。而PEAKS创新性地通过将de novo序列添加到蛋白质序列数据库并执行第二轮数据库搜索,估计FDR用于de novo肽以提高鉴定的准确性。
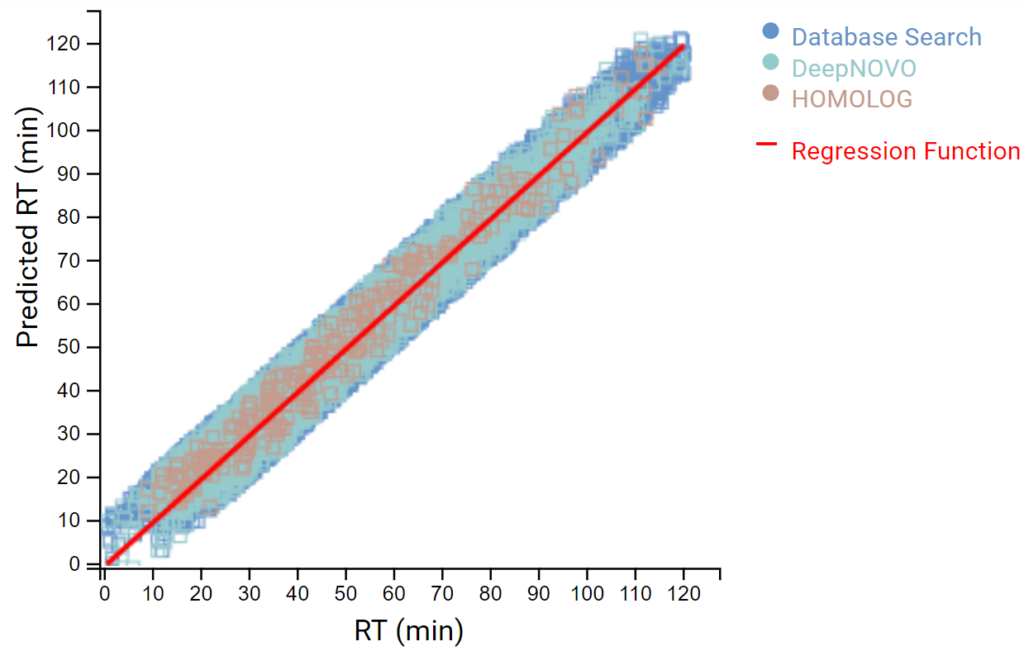
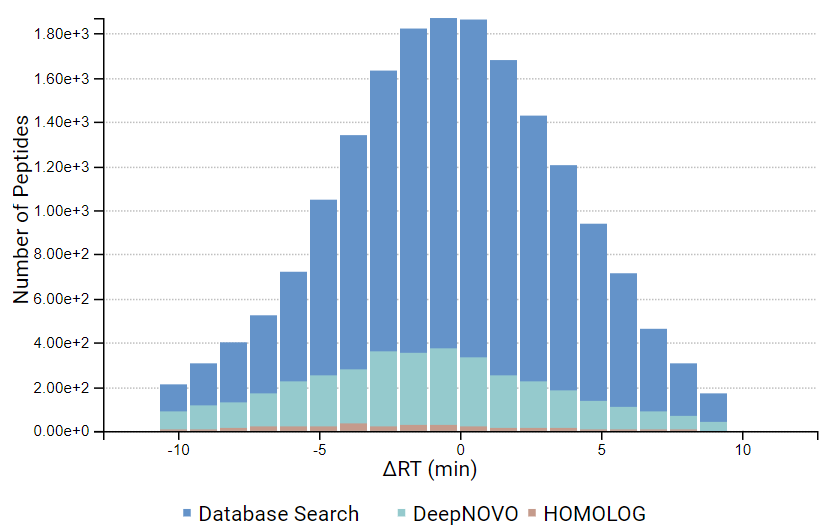
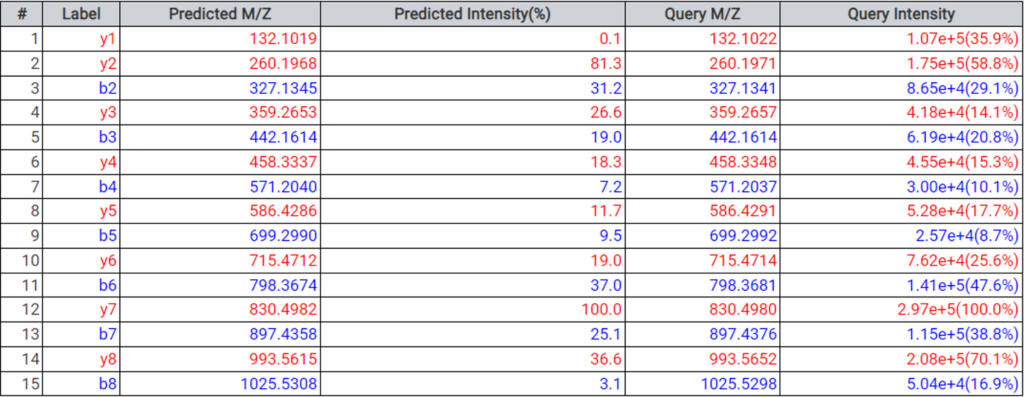
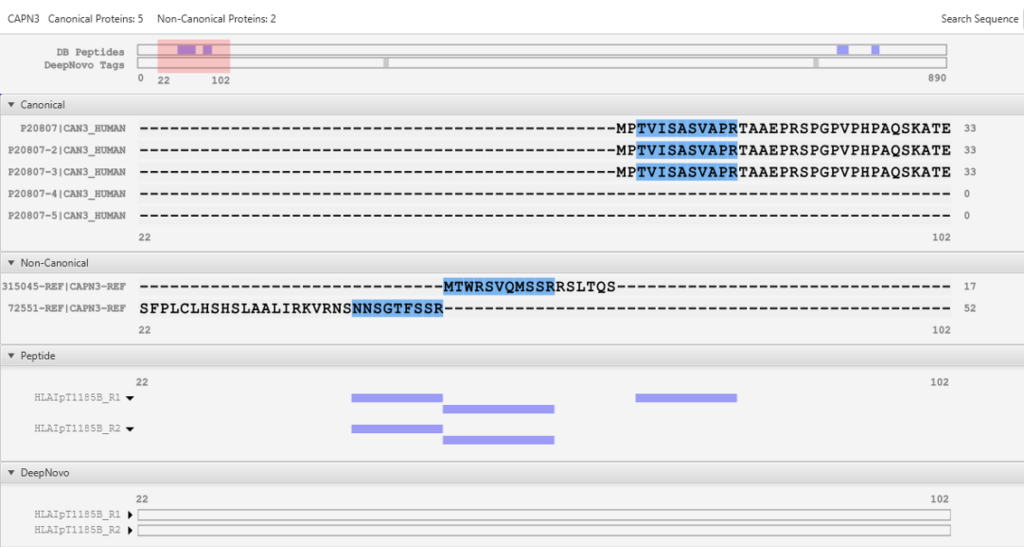
DeepNovo 多肽组工作流提供了多肽的完整鉴定列表,可以根据它们的鉴定方法进行筛选:数据库搜索,同源性搜索(具有突变的肽),或来自于从头测序的结果。结果总结页面中,用户通过统计图评估来自不同鉴定来源肽的保留时间与预测保留时间的相关性。
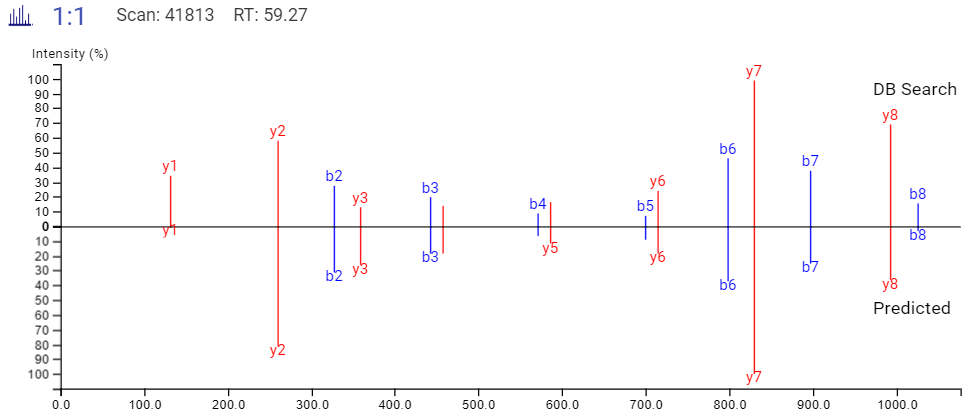
通过在结果列表中选择任意特定的肽,可以将每个谱图中碎片离子的m/z和强度值与预测的谱图进行比较。对于离子淌度数据,也预测了每个多肽的碰撞横截面积(CCS)的值。
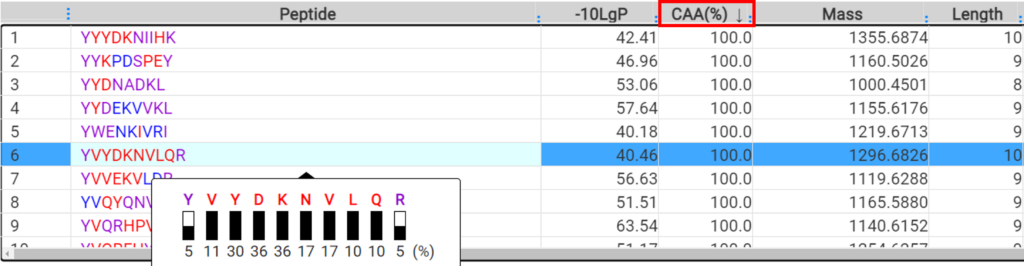
DeepNovo Peptidome工作流中添加了一种新的结果过滤方法,称为可信氨基酸百分比(confident amino acid percentage, CAA%),允许用户评估肽谱中,支持这条肽段匹配的碎片离子的信号强度值高于指定阈值的氨基酸百分比。这增加了肽序列验证的可信度。最终的多肽列表可以直接输出,用于下游分析,如结合亲和力和免疫原性预测。
DeepNovo多肽组的PTM分析
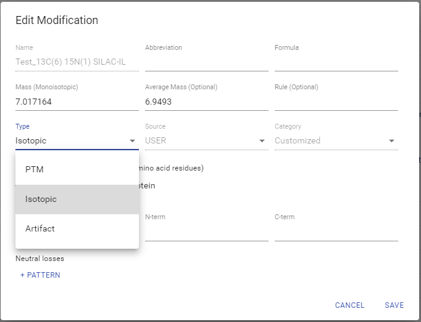
DeepNovo Peptidome工作流程现在考虑了三种不同类别的修饰,以更准确地预测修饰多肽的保留时间和MS2谱图。这些类别包括标准PTM、同位素标签和人工标记修饰(即TMT标签)。在LC-MS/MS分析之前,通常将同位素标记的肽加入免疫肽组学样品中,以验证免疫多肽的鉴定。然而,氨基酸的同位素并不像大多数生物相关的PTMs或人工标签那样影响肽的保留时间。因此,DeepNovo Peptidome工作流程中的深度学习模型被训练来预测属于这三种不同类别的肽的保留时间和MS2谱,以提高肽的评分和鉴定。
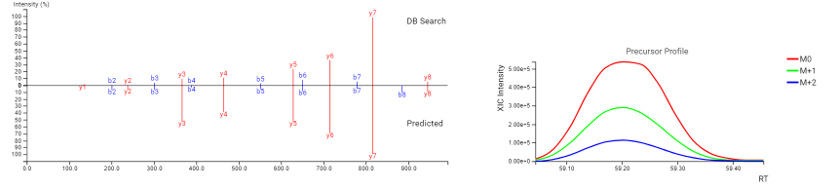
DeepNovo Peptidome DDA 工作流程拓展了传统蛋白质组学的界限
肽组学DDA工作流程旨在通过整合一个全面的gene table来弥合蛋白质组学和基因组学之间的gap。
此功能有助于多肽的鉴定与相应基因组数据之间的无缝连接。该工作流程支持non-canonical 参考序列,允许识别标准canonical 参考数据库经常遗漏的变异和修饰的序列。
对于定量分析,工作流程包括强大的label-free定量功能,并辅以自动化 QC 工具以确保数据质量和可靠性。

DeepNovo多肽组DIA数据分析
新版PEAKS 12为DIA免疫肽组学提供了一种全新的策略,集成了不依赖数据库的检索和从头测序方法[3]。获得高可信度的肽段,并根据预测的多肽的物理化学性质(包括碎片离子特征、保留时间和离子淌度等) 进行重打分,最后报告出FDR。此外,位置置信度得分与每一条肽段关联,以评估每个氨基酸的准确性。该工作流提供了一个全新的计算途径,以帮助我们在LC-MS/MS的DIA数据中鉴定由mRNA的非典型翻译或基因组变异产生的免疫肽。
Reference & Resources
References:
- Tran NH, Qiao R, Xin L, Chen X, Liu C, Zhang X, Shan B, Ghodsi A, Li M. Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nature Methods. 16(1), 63-66. 20/12/2018.
- Tran NH, Zhang X, Xin L, Shan B, Li M. De novo peptide sequencing by deep learning. Proceedings of the National Academy of Sciences of the United States of America. 114(29). 18/7/2017.
- Xin, L., Qiao, R., Chen, X. et al. A streamlined platform for analyzing tera-scale DDA and DIA mass spectrometry data enables highly sensitive immunopeptidomics. Nat Commun 13, 3108 (2022). https://doi.org/10.1038/s41467-022-30867-7
Resources
>Complete workflow for immunopeptidome analysis in PEAKS 11 (PDF)