软件操作速览
PEAKS Studio软件操作和结果解读的简要介绍(PEAKS User Manual 1.5章节). 通过使用PEAKS安装中包含的demo project,我们首先介绍PEAKS的GUI,并展示如何过滤和可视化分析结果(1-4 节). 帮助您理解如何使用PEAKS完成分析。在下面的5、6节,我们将演示如何从原始数据创建PEAKS Project并进行数据分析。
PEAKS的安装说明请参见《用户手册》的“1.3 安装与激活”。安装并运行PEAKS后,您可以通过以下两种方式之一打开示例项目(见下面的截图):
- 如果这是一个全新的安装,单击Start Page中的“Recent Project”列表中的“sample project”
- 也可以单击open project,找到PEAKS的安装目录,通过文件浏览,选择SampleProject,点击open。
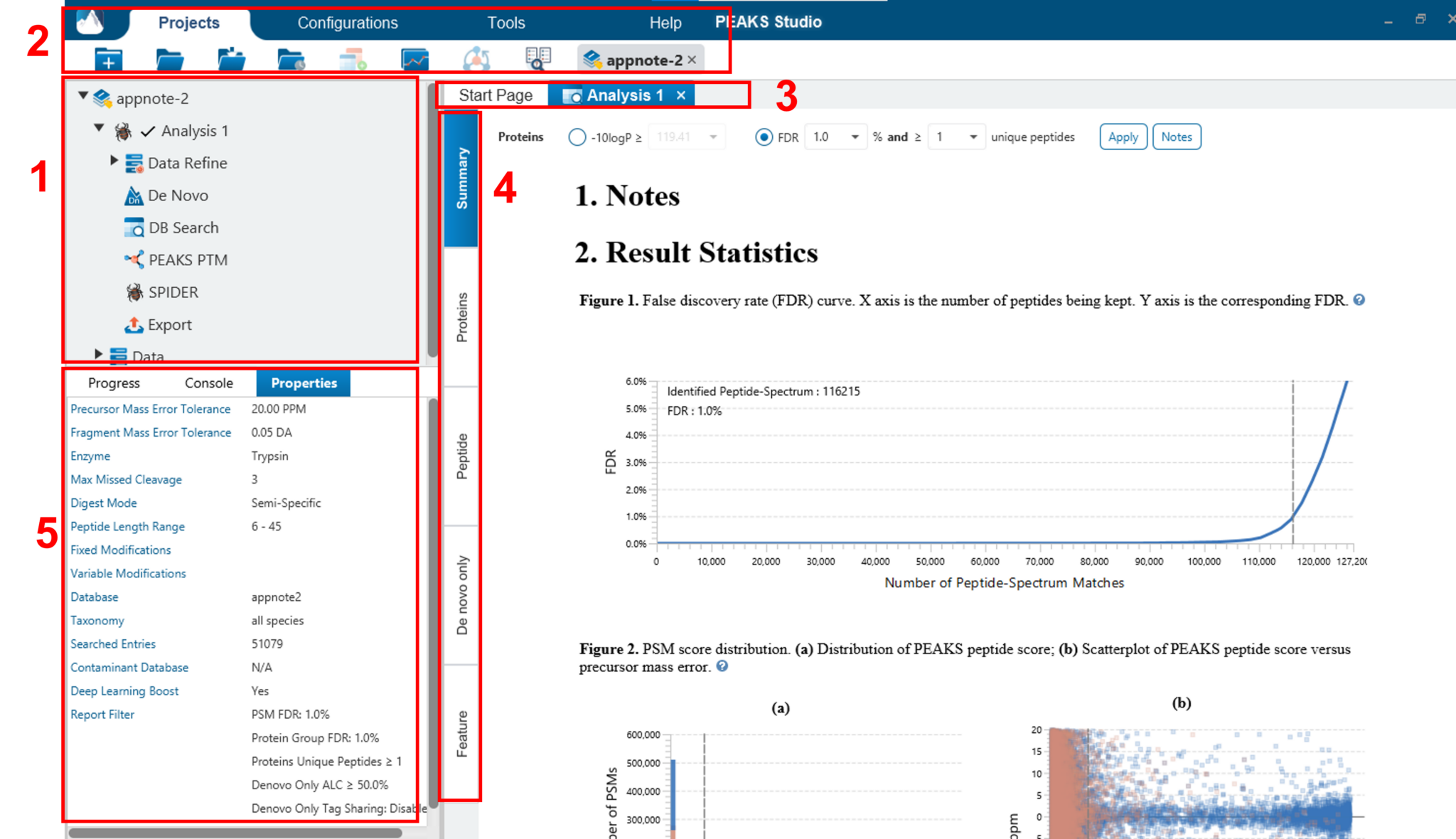
PEAKS的用户交互界面的展示以Project为导向,主要分为以下几个区域(见下图):
- 在Project树查看在该分析项目中所有的Analysis 结果节点。每一个analysis可以折叠/展开,名称可以通过右键单击anaylsis这一层次来更改,另外analysis也可以被删除。在analysis的下一层次中,可以通过双击查看在该analysis下应用PEAKS不同的功能而产生的结果节点(如Data Refine,De Novo,DB search等)以及结果导出的节点。
- 菜单和工具栏包含所有的分析工具和功能图标。通过切换顶端的标签,可分别进入Projects,Configurations,Tools和Help。在Projects标签下,可以打开/关闭Project,也可以在当前的project中新建Analysis或者修改Analysis。
- 可以通过双击Project中的结果节点来打开该节点。打开的结果节点显示在选项卡中。
- 每个打开的结果节点提供几个不同的“view” 标签。“Summary View”显示结果统计信息。Protein ,Peptide,Glycopeptide,De novo only和Feature等视图,提供数据结果的所有细节。
- Information一栏显示节点属性、运行任务进度等有用信息。
双击打开结果节点后(例如:Demo Project中PEAKS DB节点)默认显示“Summary View”,主要提供三个功能:
- 指定Protein分数阈值以过滤结果
- 检查结果统计信息
- 导出结果
“Summary View”的顶部区域是控制结果的控制面板(请参见下面的屏幕截图),底部区域是统计信息报告页面:
- 可以通过单击FDR按钮设置目标肽谱匹配的-10lgP阈值或目标FDR(错误发现率)来过滤掉鉴定结果中的低分肽。
- 通过设置目标蛋白的-10lgP的评分和特定目标肽段的计数来过滤掉鉴定结果中的低评分蛋白质。
- De Novo Only肽指的是未被数据库搜索算法识别的具有可信的De Novo序列标签的肽。要报告De Novo Only肽,ALC(平均局部置信度)评分必须等于或优于指定的阈值。同时,频谱的最佳数据库搜索结果的分数不应大于指定的 -10lgP 阈值。
- 默认情况下,用于De Novo Only的-10logP阈值被锁定为与用于筛选肽的-10lgP阈值相同。要指定其他值,只需单击锁定图标即可解锁筛选程序。
更改筛选条件后,筛选程序将更改为红色。单击它以应用新条件。

顶部控制窗格有两个附加按钮:“Export”和“Notes”。单击“Export”将显示多个选项和文件格式,用户可以使用这些选项和文件格式导出分析结果。单击“Notes”允许用户添加有关项目的文本注释,该注释将显示在“Summary”视图页面的顶部和HTML导出中。
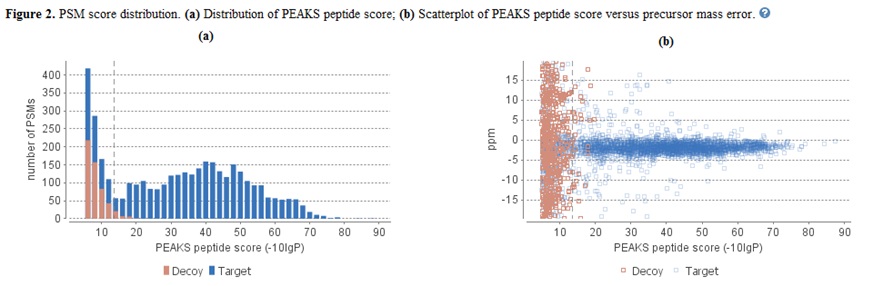
应用筛选条件后,“Summary”视图底部的统计信息报告页面将相应更新。这里只解释了两个统计图表(见下面的屏幕截图)。
图2(a)显示了堆叠直方图中的PSM分数分布。如果搜索结果和肽-10lgP分数阈值都具有高置信度,则在高分区域中应观察到较少的假目标匹配(棕色)。此外,如果FDR估计方法(诱饵融合)工作正常,则相对于目标(蓝色)匹配,应在低分区域中观察到相似或更多数量的假目标匹配(棕色)。
图2(b)绘制了所有PSM的母离子质量数误差与-10lgP肽评分的关系。该图对于高分辨率仪器最有用。通常,高分点应以质量误差 0 为中心。请注意,当数据点低于某个分数阈值时,它们开始分散到更大的质量误差。
除“Summary View”外,PEAKS还将结果分为五个部分:“Feature”、“Protein”、“Peptide”、“De Novo Only”和“LC/MS”。
- “Feature View”包含通过筛选程序的所有可检测特征的列表。每个feature都将列出与其关联的所有 MS/MS 扫描。
- “Protein View”包含通过筛选程序鉴定的蛋白质列表。用同一组(或肽的子集)鉴定的蛋白质被分组在一起。
- “Peptide View”显示通过筛选程序鉴定的所有肽。鉴定相同肽序列的多个光谱被分组在一起。
- “De Novo Only View”显示了仅通过De Novo测序鉴定出的所有肽。
- “LC/MS View”将 LC-MS 数据显示为热图,突出显示 MS/MS 扫描和检测到的特征。
在这里,重点将放在蛋白质覆盖视图上。单击“Protein View”选项卡,然后选择一种蛋白质。相应的蛋白质覆盖图将显示在“Protein View”的底部。蛋白质覆盖视图将选定蛋白质的所有鉴定出的肽映射到蛋白质序列上。它能够毫不费力地检查每个PTM和每个氨基酸的突变。下面列出了蛋白质覆盖率视图上一些最常用的操作(请参见下面的屏幕截图):
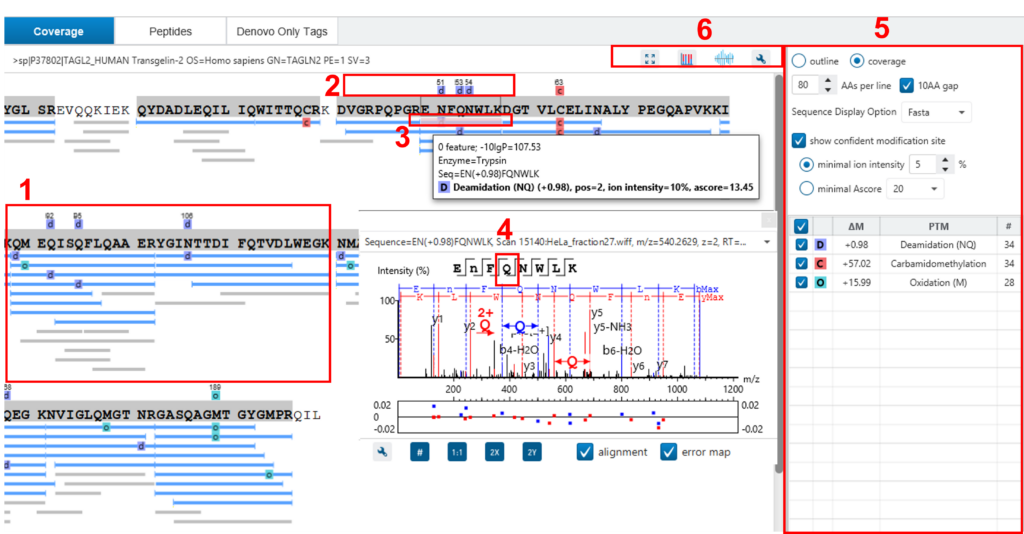
- 每个蓝色条表示已鉴定的肽序列。灰色条表示仅从头标记匹配。
- PTM和突变用彩色图标和白色信箱突出显示。高度可信的PTM和突变显示在蛋白质序列的顶部。
- 单击肽可显示光谱注释。
- 将鼠标悬停在氨基酸上可显示支持碎片离子峰。
- 用于控制覆盖视图显示的选项。
- “coverage/outline”选项可以显示或移除肽条。
- “sequence display option”可以通过快速显示或在项目设置期间选择的酶选项来突出显示覆盖图。
- “de novo tags sharing”指定De Novo Only序列与蛋白质之间连续氨基酸匹配的最小数量,然后才能显示为灰色条。
- “de novo peptides fully matched”复选框允许在序列(无论其长度如何)与蛋白质中的序列完全匹配时显示从头肽。
- “minimum ion intensity”指定在PTM位置被视为置信并显示在蛋白质序列顶部之前,其中一个MS / MS谱图中的最小碎片离子相对强度。
- PTM列表中的复选框允许用户查看结果中具有特定PTM的肽。单击彩色框可更改颜色。双击 PTM 名称以查看 PTM 详细信息。
- 全屏按钮、PTM 图谱按钮、肽图分析按钮和工具箱按钮:
要从原始数据文件创建新的PEAKS项目,请执行以下步骤(请参见下面的屏幕截图):
- 选择“新建项目…或单击工具栏上的“新建项目”
 图标。将出现“Project Wizard”。
图标。将出现“Project Wizard”。 - 使用“添加数据”按钮添加要加载的所需文件,然后单击“打开”。所有选定的数据文件都将列在左侧。
- 将列表中的选定数据放入样本中:用
 将所有文件放入新样本中; 用
将所有文件放入新样本中; 用 将它们放入现有样本中; 或者使用
将它们放入现有样本中; 或者使用 将它们放入每个文件的单独样本中;如果要按分隔符或正则表达式对文件进行分组,可以单击该按钮
将它们放入每个文件的单独样本中;如果要按分隔符或正则表达式对文件进行分组,可以单击该按钮  。
。 - 点击
 或
或 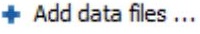 按钮可分别向项目添加示例或将数据文件添加到示例
按钮可分别向项目添加示例或将数据文件添加到示例 - 对于每个样品,指定样品详细信息:“仪器”类型、“碎片”方法、“酶”名称和数据“采集”。
- 注意:用户可以为每个样品指定不同的蛋白水解酶。使用多种酶分析相同的蛋白质可以产生重叠的肽,这将增加蛋白质覆盖率。
- 注意:要将相同的示例详细信息应用于整个项目,请选择具有正确设置的示例,然后单击“复制到整个项目”按钮。
- 单击“完成”按钮以创建项目
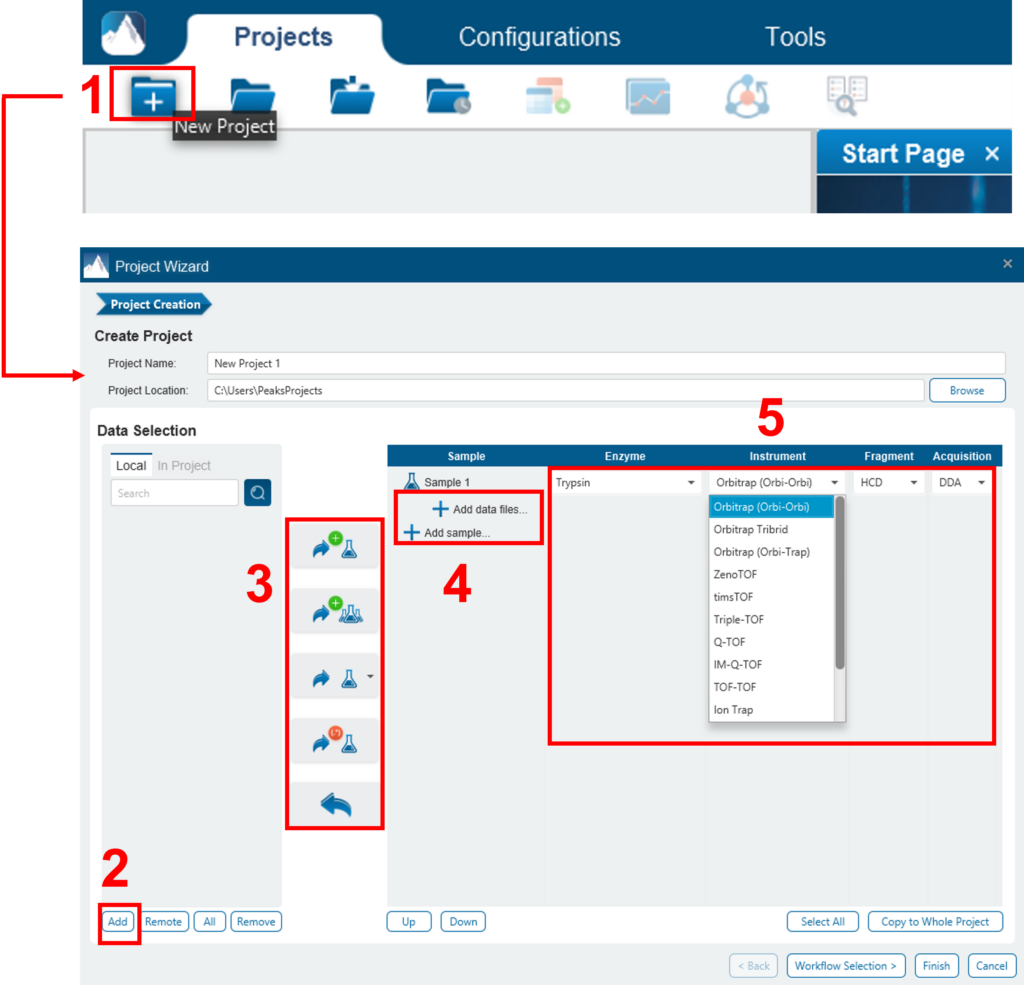
进行分析:1) 在项目视图中选择项目、样本或结果节点;2) 单击所需的分析工具按钮。在以下示例中,将从“Project View”窗格中选择完成的De Novo结果,并将使用这些结果进行 PEAKS DB 搜索。PEAKS DB是执行数据库标识搜索的工作流。
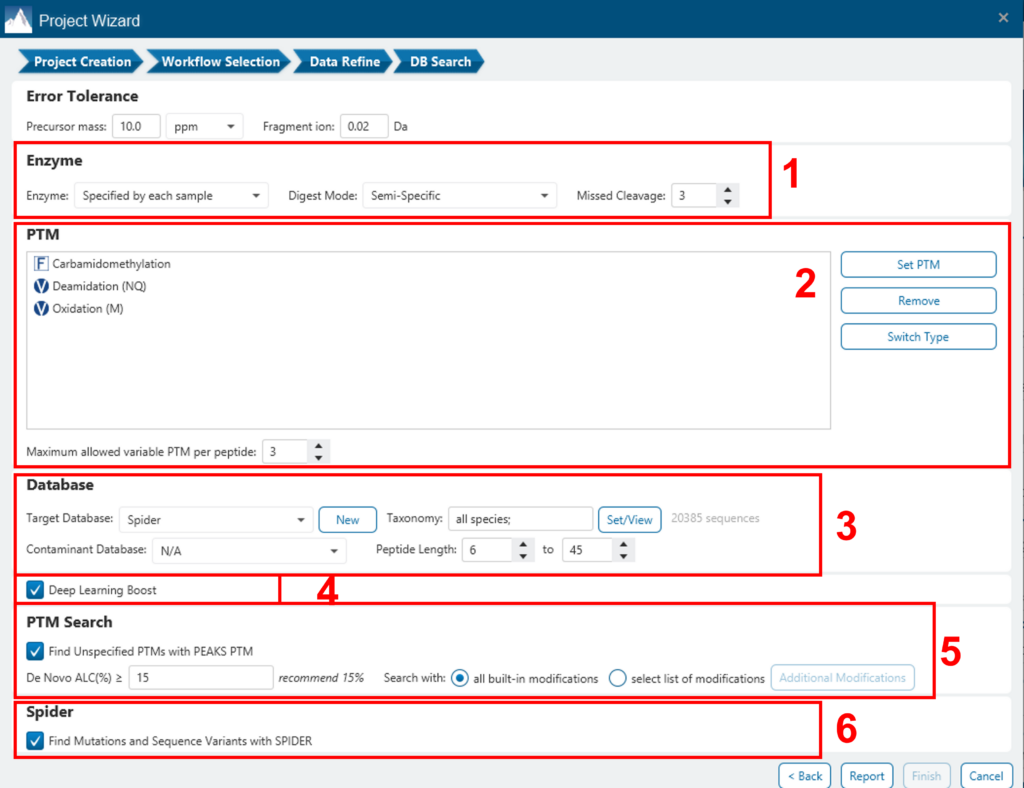
将出现一个窗口,允许用户指定所需的分析搜索参数。PEAKS DB的大多数搜索选项都是标准且直接的。下面提供了更多详细信息:
- 如果在项目创建步骤中为每个样品指定了蛋白水解酶,用户可以选择使用每个样品中已指定的酶。这使得在单个项目和单个搜索中使用多种酶成为可能。
- 指定样本中预期的固定 PTM 和一些常见的可变 PTM。
- 选择蛋白质序列数据库,或复制并粘贴蛋白质序列以进行数据库搜索。用户还可以选择要包含在数据库搜索中的污染物数据库(可选)。
- 使用相同的参数进行从头测序,或基于现有的De Novo测序结果节点进行搜索。
- 使用诱饵融合方法估计错误发现率 (FDR)。
- 诱饵融合是一种增强的目标诱饵方法,用于使用 FDR 验证结果。诱饵聚变将诱饵序列附加到每个蛋白质作为搜索的“阴性对照”。有关更多详细信息,请参阅 BSI 的网络教程。
- 在 PEAKS DB 数据库搜索完成后启用 PEAKS PTM 和 SPIDER 算法。
- 默认情况下,PEAKS PTM将执行额外的PTM搜索,该搜索会考虑Unimod的所有313个自然发生的修改。用户可以通过单击“高级设置”按钮来指定他们希望用于其他PEAKS PTM搜索的PTM的自定义列表。
- SPIDER基于De Novo测序标签进行同源搜索。如果选择,SPIDER算法将在每个置信度的De Novo标签(ALC>15%)上进行,这些标签的频谱未被PEAKS DB以高置信度(-10lgP < 30)识别。SPIDER将通过改变数据库肽的氨基酸来构建新的肽序列。对于每个谱图,由SPIDER构建或由PEAKS DB发现的更好的序列将用作鉴定的肽。SPIDER适用于跨物种搜索和查找蛋白质的点突变。可以通过此工作流调用 SPIDER,也可以通过单击工具栏中的 SPIDER 图标来调用。