错误发现率(FDR)
PEAKS FDR 评估
概览
从质谱数据中鉴定多肽通过软件实现自动化。然而,就像科学实验需要使用对照进行一样,软件的肽鉴定结果也需要经过统计验证以避免假阳性。对于当今的肽鉴定,最被广泛接受的结果验证方法是错误发现率(FDR)。 这篇文章解释了什么是FDR;它是如何实际运算的;以及使用FDR控制中的一些常见错误。
每个肽鉴定软件的核心功能都是对肽和 MS/MS 谱的匹配质量的评估。对于数据中的每张MS/MS谱图,软件都会搜索蛋白质数据库,以找到最高肽谱匹配分数的肽。谱图与得分最高的肽之间的匹配通常称为肽-谱匹配(peptide-spectrum match,PSM)。
一系列原因可能导致肽谱匹配的错误,其中包括:
- 低质量的谱图;
- 肽不在数据库当中
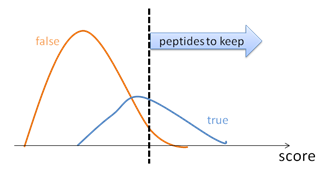
- 打分体系不够完善。为了控制结果质量,PSM 按其得分来排序。通过选择合适的分数阈值,可以得到满足高于阈值条件质量的PSMs(图1)。错误发现率FDR指的是错误的PSMs 和在阈值之上的PSMs总数的比率。
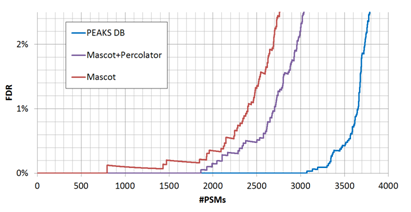
通过调整阈值,结果的准确度(FDR) 可以与灵敏度(鉴定到的数量)进行转化。软件不同,其评分体系的,可能具有显著不同的权衡效率,如 图 2中的FDR曲线所示。
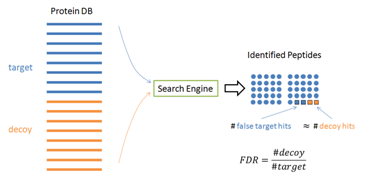
在实践当中,很难分辨哪个PSM是错误的—否则这些错误的PSMs可以被算法删除以实现零错误率。 因此,Target-Decoy方法[1]在实践中被广泛用于估算FDR。在这个方法当中,软件会在相同大小的target数据库和decoy数据库进行搜索。 如果decoy库建的是足够准确的,那么软件鉴定出的错误应当在target库和 decoy当中均匀分布。由于所有的decoy中得到的鉴定都应当是错误的,FDR 可以通过 FDR = (# Decoy hit) / (# target hit)来估计。
Target-Decoy方法使用的常见误区
如果使用得当,Target-Decoy方法在统计学上是估算FDR的合理方法。不过,这种方法的误用很常见,并且会导致对结果质量的过高评估。在这里,我们简要总结一些常见的错误。需要强调的是,在上一节中讲到的“相同大小”和“均匀分布”是正确使用Target-Decoy方法的前提条件。我们即将看到,大多数Target-Decoy方法的使用错误都是由于违反了这些先决条件。
错误 1
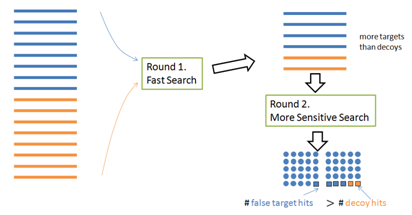
使用方法Target-Decoy来验证搜索软件中的多轮搜索方法。
为了加快搜索速度,多轮搜索算法通常在第一轮从大型数据库中选择一个蛋白质的候选列表,然后在第二轮的蛋白质候选列表(而不是整个数据库)中鉴定到更多的PSM。然而,这种方法使用Target-Decoy法将无效:在第一轮中选择的目标蛋白多于Decoy蛋白数目;因此,在第二轮中,target和decoy的大小不同(图4)。
错误 2
使用Target-Decoy方法时,通过蛋白鉴定信息,来反馈给搜索软件中的肽谱匹配进行奖励性加分。
一个蛋白的PSM越多,代表这个蛋白的置信度就越高。 因此,许多软件工具会给来自高置信度蛋白的肽加分。尽管这样做可以提高搜索的灵敏度,却让Target-Decoy方法变得不准确:会出现更多具有高分的Target蛋白的匹配;因此,因高分带来的错误的target蛋白匹配将会比decoy错配获得的蛋白更多。错误匹配将不会均匀分布。
错误 3
通过应用Target-Decoy方法时,用重新训练出的模型来进行对结果的重排。
这种结果重排的策略最近被越来越广泛的使用,因为它可以提高搜索的灵敏度。 然而,这也会令Target-Decoy方法变得不准确:一个较为粗放的重新学习算法会用到过多的参数,使得数据出现过度拟合并消除decoy hit(但并不是target库中的错配)。因此,这种策略仅适用于当重新训练算法的设计考虑了过拟合问题,并且数据集非常大的情况。
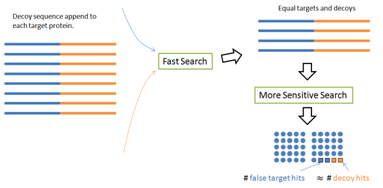
诱饵融合方法
有一个简单的改进可以避免前两个常见错误——PEAKS DB的论文[1]提出了一种decoy- fusion的方法。decoy- fusion方法不是将target和decoy数据库连接在一起,而是将同一蛋白质的target序列和decoy序列连接在一起,作为“fusion”序列(图5)。这个简单的更改会产生一些有意义的不同。对于两轮搜索问题,第二轮的target和decoy长度仍然相同。对于蛋白质奖励性得分问题,相同数量的给分将同样的应用于同一融合序列的target和 decoy部分。 因此,“相同大小”和“均匀分布”的先决条件被重新创建;FDR值能够被重新准确的估算。PEAKS软件的内置结果验证正是使用的这种decoy- fusion方法。
Zhang J, Xin L, Shan B, Chen W, Xie M, Yuen D, Zhang W, Zhang Z, Lajoie G.A., Ma B, PEAKS DB: De Novo Sequencing Assisted Database Search for Sensitive and Accurate Peptide Identification. Mol. Cell. Proteomics. 11, M111.010587 (2012).
Xin, L., Qiao, R., Chen, X. et al. A streamlined platform for analyzing terascale DDA and DIA mass spectrometry data enables highly sensitive immunopeptidomics. Nat Commun 13, 3108 (2022). doi:10.1038/s41467-022-30867-7